Introduction
Lung cancer causes cancer-related deaths worldwide [1]. Recent advances in the understanding of oncogenic drivers and the development of targeted agents have improved the survival of patients with non–small cell lung cancer (NSCLC). Among the frequently occurring driver oncogenes in NSCLC, epidermal growth factor receptor (EGFR) mutations are the most common genetic alterations in Korean patients, especially those with lung adenocarcinoma [2]. In patients with NSCLC with activating EGFR mutations, first-generation EGFR tyrosine kinase inhibitors (TKIs) greatly improve the response rate and survival. However, most patients acquire resistance to EGFR-TKIs [3,4]. Cytotoxic chemotherapy is generally the treatment of choice in patients who have not acquired the EGFR T790M mutation after resistance to first- or second-generation EGFR-TKIs, or in patients who have failed third-generation EGFR-TKIs [5]. However, in these settings, the response rate to chemotherapy is only 20%–30%, and progression-free survival (PFS) is 3–6 months [6,7]. Therefore, a more effective therapeutic strategy is urgently required for these patients.
In normal tissues, adenosine originates from extracellular adenosine triphosphate and is involved in anti-inflammatory, cytoprotective, and wound-healing processes [8–10]. CD73 (ecto-5′-nucleotidase, NT5E) exists on the surface of various cells such as endothelial, immune, and stromal cells, serving as an enzyme that hydrolyzes adenosine monophosphate to adenosine [11]. CD73 is frequently overexpressed in various cancers, including lung, breast, ovarian, colorectal, and prostate cancers [12,13]. High expression of CD73 in tumor cells is correlated with poor survival in many cancers [12,13]. Focusing on the immunosuppressive function of the adenosine pathway, its inhibition has been studied as a potential target for immunotherapy in various cancers, including EGFR-mutant NSCLC [14–16].
Recently, there has been growing evidence that CD73 plays an important role in tumor growth and survival, independent of its enzymatic activity [13,17–19]. Several studies have shown that CD73 is involved in cancer proliferation through the EGFR signaling pathway [17,18,20]. A previous study revealed that the expression of CD73 is controlled by miR-30a-5p, and CD73 plays a role in promoting tumor growth through EGFR signaling in EGFR wild-type lung cancer cell lines [18]. However, the role of CD73 in EGFR-mutant NSCLC remains unclear.
Thus, this study aimed to investigate whether targeting CD73 could be a potential therapeutic approach for EGFR-mutant NSCLC patients who have developed acquired resistance to EGFR-TKIs.
Materials and Methods
1. Patients and tumor samples
We obtained paired tumor tissues from 26 patients with metastatic EGFR-mutant NSCLC before and after they developed acquired resistance to first-generation EGFR-TKIs at Seoul National University Hospital (SNUH). The clinicopathological data were retrospectively reviewed.
2. Immunohistochemistry
Immunohistochemistry (IHC) staining was performed on 4-μm-thick sections obtained from whole tissue blocks. All slides were treated to remove wax and were rehydrated in a graded series of alcohol solutions. IHC staining was performed using BenchMark ULTRA (Ventana Medical Systems, Tucson, AZ). Anti-CD73 antibody (#13160, clone D7F9A, rabbit monoclonal antibody, Cell Signaling Technology, Danvers, MA) was diluted 1:100. After heat-induced antigen retrieval, samples were incubated with primary antibodies for 32 minutes. Binding of the primary antibody was detected using the OptiView DAB IHC detection kit (Ventana Medical Systems) according to the manufacturer’s instructions. CD73 IHC was performed based on the intensity and proportion of membranous staining. The staining intensity was scored from 0 to 3 (0, absent; 1, weak; 2, moderate; 3, strong). The H-score was determined by multiplying the staining intensity by the percentage of positive cells, ranging from 0 to 300. Programmed death-ligand 1 (PD-L1) IHC using rabbit anti–PD-L1 (E1L3N) XP monoclonal antibody (Cell Signaling Technology) was performed and assessed as previously described [21].
3. Cell lines and reagents
EGFR exon 19 E746-A750 deletion (19del) mutant lung cancer cell line PC9 was purchased from Riken BioResource Research Center (Tokyo, Japan). EGFR-TKI-resistant cell lines (ER and GR) were established by exposing parental PC9 cells to erlotinib and gefitinib, respectively, at a dose escalation of 100 nmol/L and 1 μmol/L of each inhibitor. PC9ER and PC9GR retained EGFR 19del but did not acquire the T790M mutation [22]. The subclonal-resistant cell lines exhibited ≥ 5-fold greater IC50 values for EGFR-TKIs than the parental cells, as determined using the cell viability assay, and this phenotype was stable for at least 6 months without EGFR-TKIs treatment.
All cell lines were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA) and 1% penicillin/streptomycin (Gibco). The EGFR-TKIs gefitinib (Iressa) and erlotinib (Tarceva) were purchased from Selleck Chemicals (Boston, MA). Antibodies against total EGFR (#4267S), phospho-EGFR (#3777S), total AKT (#4685), phospho-AKT (#4060S), total ERK 42/44 (#9102), phospho-ERK (#9106), p21 (#2946), cyclin D1 (#2926), and glyceraldehyde 3-phosphate dehydrogenase (#5174) were purchased from Cell Signaling Technology.
4. Short heparin RNA transfection
Short heparin RNA (shRNA) against CD73 and the negative scrambled control were cloned into packaging plasmids (ORIGENE, Rockville, MD). Lentiviruses were generated using the packaging cell line HEK293T purchased from the American Type Culture Collection (ATCC). The viral supernatant was harvested at 48-hour post-transfection. After passing through 0.45 μmol/L syringe filters, the appropriate amount of virus was used to infect target cells in the presence of 8 μg/mL polybrene. The sequences of shCD73-A was 5′-GTCTATCTGGATGGCTCC-TCTCAATCATG-3′, and sh- CD73-B was 5′-GGCGGAATCCATGTGGTGTATGATCTT-TC-3′. Cells infected with viruses underwent selection with 1 μg/mL of puromycinPC9ER-shCD73 and PC9GR-shCD73 refer to each cell stably transfected with shRNA against CD73, respectively. NC refers to cells stably transfected with the vector alone as a negative control.
5. mRNA extraction and quantitative real-time polymerase chain reaction
Total RNA was isolated from the cells using an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Each sample was reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). cDNA was prepared as previously described. Quantification of gene expression levels and an internal reference gene (β-actin) was performed using the SYBR green detection method. The Power SYBR Green PCR Master Mix (Life Technologies, Warrington, UK) was used according to the manufacturer’s instructions. Relative gene expression was calculated using the ΔΔCt method with β-actin as the internal reference gene. Cycling conditions were 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds, 60°C for 30 seconds, and 72 °C for 30 seconds. The Applied Biosystems StepOnePlus Real-Time PCR System (Life Technologies) was used for cDNA amplification and analysis. The nucleotide sequences of the primers used were as follows: CD73-F (5′-GAA CCT GGC TGC TGT ATT GC-3′) CD73-R (5′-ACA CCA CAT GGA TTC CGC C-3′).
6. Flow cytometry and apoptosis analysis
CD73 expression in cell lines was measured by flow cytometry using fluorescence-conjugated mouse monoclonal anti-CD73 antibodies from eBioscience (clone AD2). The cell lines treated with EGFR-TKIs or vehicle were stained with Annexin V and propidium iodide (PI; BD Pharmingen, San Jose, CA), and the cells were analyzed by flow cytometry using a FACSCalibur instrument (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software ver. 7.6 (FlowJo, LCC).
7. Immunoblot assay
Cells were plated in 6-well plates and treated with EGFR-TKIs at 100 nmol/L and 1 μmol/L for 6 or 24 hours. Subsequently, cell lysis was performed with diluted 10× cell lysis buffer (Cell Signaling Technology, Danvers, MA), phenylmethanesulfonylfluoride (Sigma-Aldrich, St. Louis, MO), PhosSTOP (Roche, Basel, Switzerland), and proteinase inhibitor cocktail (Merck, North Wales, PA), and quantified with protein assay dye reagent concentrate (Bio-Rad, Hercules, CA). The prepared samples were separated using NuPAGE Bis-Tris Gels (Invitrogen), transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad), and detected using ECL Prime Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK). Image analysis was performed using an Image Quant LAS-4000 mini (GE Healthcare).
8. Cell proliferation and viability assays
Cells (1×105) were plated in 6 cm2 dishes and cultured. Cells were harvested by trypsinization, stained with Trypan blue, and counted using a LUNA Automated Cell Counter (Logos Biosystems, Anyang, Korea) on days 2, 5, 6, and 7. Three independent experiments were performed. Cell proliferation curves were plotted based on average cell counts.
For the CellTiter Glo (CTG) cell viability assay, cells were seeded in a 96-well plate at 3,000–5,000 cells/well and incubated overnight. The cells were then cultured in the presence of each EGFR-TKIs or vehicle for 72 hours. Cell proliferation was analyzed using the CTG-luminescent cell viability assay (Promega, Madison, WI), and the luminescent signal was measured using a GloMax Navigator Microplate Luminometer (Promega). Concentrations that inhibited 50% (IC50) values and graphs were determined using SigmaPlot 12.0 software (Systat Software Inc., San Jose, CA) and GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA). These experiments were independently repeated three times.
9. Cell cycle analysis
Cell cycle distribution was determined using PI staining. Cells were seeded at a density of 2×105 cells/well in 6-well plates. After incubation for 16 hours, the cells were treated with 1 μmol/L EGFR-TKIs for 72 hours and then harvested. Following treatment, the cells were washed with DPBS, fixed in 75% cold ethanol, and incubation for at −20°C overnight. Fixed cells were washed in phosphate buffer saline (PBS) and resuspended in 150 μL PBS. A total of 5 μL PI (1 mg/mL) and 10 μL of RNase A (10 mg/mL; Sigma-Aldrich) were added to a 150 μL cell suspension, which was then gently vortexed at room temperature. The cell cycle was analyzed using a FACSCalibur flow cytometer (BD Biosciences) and ModFit LT (Verity Software House, Topsham, ME).
10. Colony-forming assay
Cell lines were plated into 12-well plates and treated with EGFR-TKIs for 14 days, with the media changed every 2–3 days. The starting cells were established at 90% confluence in the control group on day 14. Colonies were washed with PBS, fixed in absolute ethanol for 1 hour, and stained with 0.1% Coomassie Brilliant Blue-R250. Images were captured using an EVOS Cell Imaging System (Thermo Fisher Scientific, Waltham, MA).
11. Statistical analysis
All analyses were performed using SPSS ver. 25 (IBM Corp., Armonk, NY), GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA), and SigmaPlot ver. 12.0 (Systat Software Inc., San Jose, CA). Kaplan-Meier estimates were used for both overall survival (OS) and PFS analysis. Survival was compared using the log-rank test. Pearson’s chi-squared test was used to compare categorical data between the two groups according to CD73 protein expression. A two-tailed t test was used to determine significant differences among the groups. All p-values were two-sided, with p < 0.05 indicating statistical significance.
Results
1. CD73 expression and survival in EGFR-mutant NSCLC
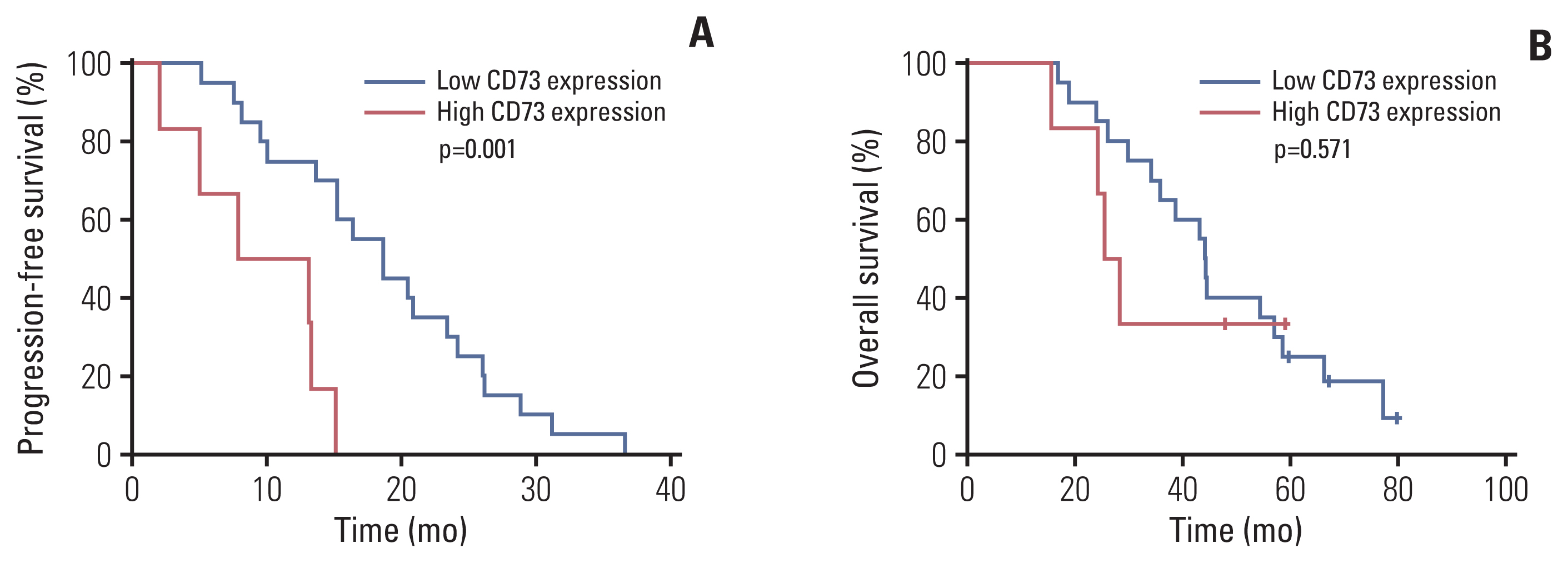
Paired tumor tissues before and after resistance to first-generation EGFR-TKI treatment were obtained from 26 patients with metastatic EGFR-mutant NSCLC. Baseline characteristics according to CD73 protein expression in pretreatment tumor samples are shown in Table 1. The range of H-scores for the tumors was 0 to 240. Among tumors showing moderate to high intensity, those with an H-score of 60 or higher were defined as the high-expression group, while the rest were defined as the low-expression group. All patients’ tumors acquired resistance to the first-generation EGFR-TKIs with median PFS of 15.2 months (range, 1.9 to 36.7 months). Patients with high expression of CD73 had significantly worse PFS than patients with low expression of CD73 (median PFS, 7.8 vs. 18.6 months; p=0.001) (Fig. 1A), and the OS also showed a similar trend toward poorer prognosis without statistical significance (median OS, 25.4 vs. 44.3 months, p=0.571) (Fig. 1B). Therefore, CD73 expression is a poor prognostic factor in patients with EGFR-mutant NSCLC.
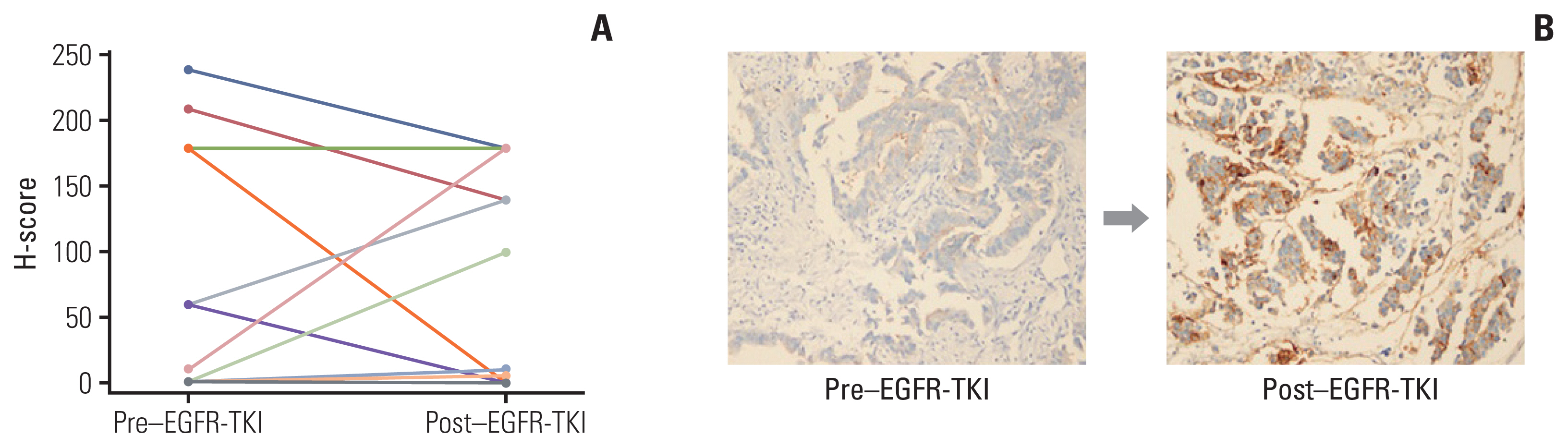
When comparing CD73 protein expression before and after resistance to EGFR-TKIs, CD73 protein expression did not change in 18 of 25 patients, but increased in seven patients (26.9%) and decreased in three patients (11.5%) (Fig. 2A). Representative IHC staining results for CD73 protein expression are shown in Fig. 2B.
2. Effect of CD73 silencing on in vitro cell proliferation and EGFR-TKI resistance
We used shRNA targeting CD73 to silence CD73 in EGFR-mutant lung cancer cell lines. The mRNA and protein expression of CD73 was determined by quantitative real-time polymerase chain reaction, flow cytometry, and immunoblot analysis to further confirm the specificity of shRNA-mediated silencing of CD73 (S1A–S1C Fig.).
We assessed the effects of CD73 silencing on in vitro cell proliferation. The cell proliferation curve showed cell proliferation in the shCD73 group (S1D Fig.). To determine whether CD73 inhibition contributes to the reversal of EGFR-TKI resistance, we performed a cell viability assay for 72 hours. PC9ER-shCD73 and PC9GR-shCD73 cells showed a significant and dose-dependent decrease in cell viability following treatment with erlotinib and gefitinib, respectively, whereas NC cells showed no difference in cell viability following EGFR-TKI treatment (PC9ER group, IC50 > 5 vs. 0.0210±0.001 nmol/L, p < 0.001; PC9GR group, IC50 > 10 vs. 0.0063±0.0033 nmol/L, p < 0.001) (Fig. 3A). We observed that in the PC9 cell line, CD73 inhibition did not affect sensitivity to EGFR-TKI treatment (S2 Fig.). We performed a long-term colony formation assay using Coomassie Brilliant Blue-R250. Compared with NC cells, 14-day colony formation was significantly decreased in PC9ER-shCD73 and PC9GR-shCD73 cells treated with various concentrations of erlotinib or gefitinib (Fig. 3B). Taken together, sensitivity to EGFR-TKI was restored through CD73 inhibition, and this effect was long-lasting.
The inhibitory effects of EGFR-TKI in PC9ER-shCD73 and PC9GR-shCD73 cells were further investigated using an immunoblot assay. A greater decrease in phospho-EGFR expression was observed in a dose-dependent manner in PC9ER-shCD73 and PC9GR-shCD73 cells than in NC cells (Fig. 3C). This result suggests that the EGFR signaling pathway may have been involved in the inhibition of cell growth by CD73 inhibition.
3. Cell cycle arrest and apoptosis through CD73 inhibition and EGFR-TKI
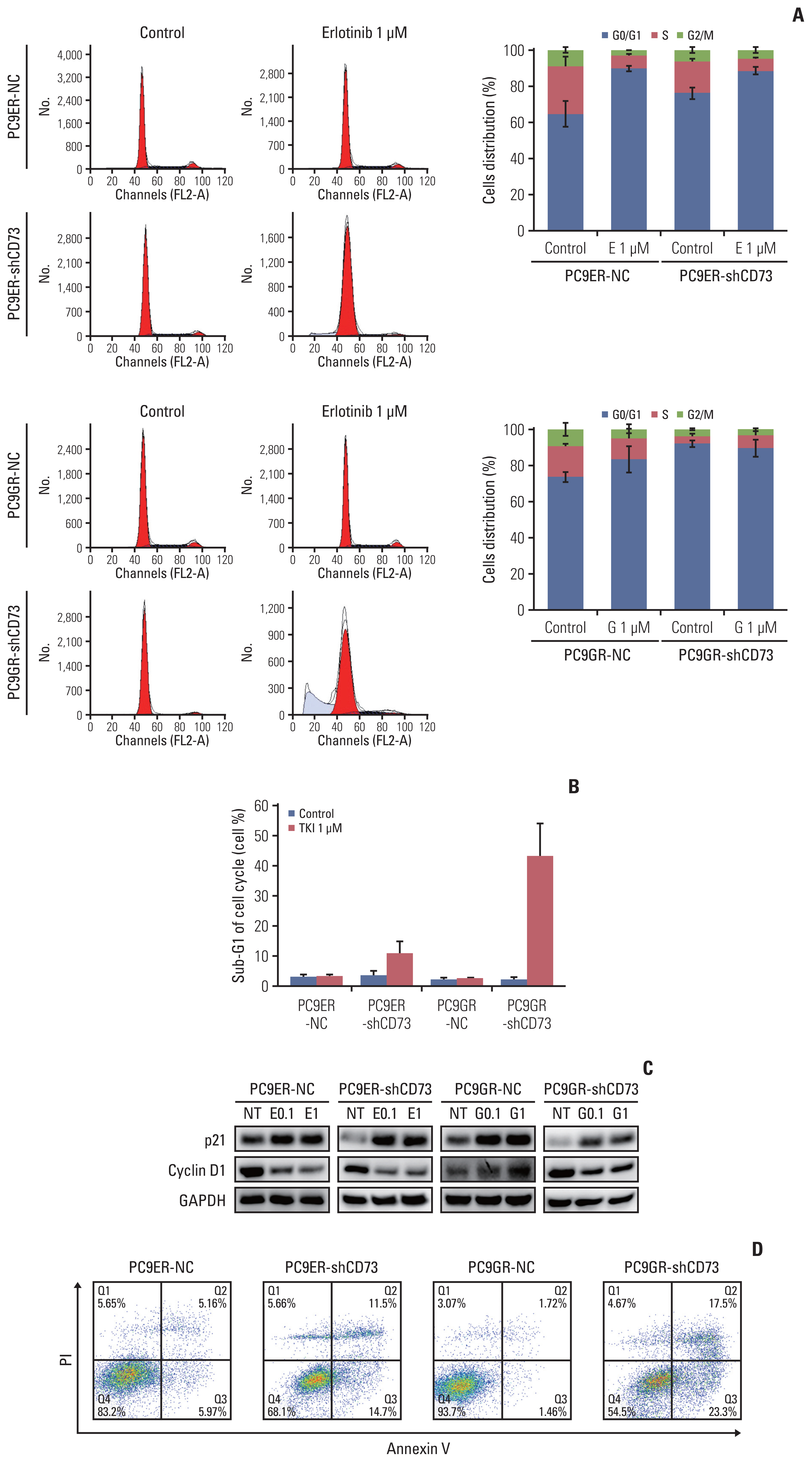
The effects of CD73 silencing on cell cycle progression and apoptosis were determined using flow cytometry. In the control group, the proportion of G0/G1 cells increased in PC9ER-shCD73 and PC9GR-shCD73 cells compared to NC cells. When treated with EGFR-TKI, the proportion of G0/G1 cells was similar between PC9ER-NC and PC9ER-shCD73 cells, while an increase in the proportion of G0/G1 cells was observed in PC9GR-shCD73 cells compared to PC9GR-NC cells. This finding provides a possible mechanism whereby CD73 inhibition could induce growth inhibition by affecting cell cycle arrest in the G0/G1 phase (Fig. 4A). We examined cell cycle-related protein expression in each cell using an immunoblot assay. When PC9ER-shCD73 and PC9GR-sh-CD73 cells were treated with EGFR-TKIs, increased expression of p21 and decreased expression of cyclin D1 were observed, which could explain G0/G1 arrest (Fig. 4C).
The increased sub-G1 fractions in PC9ER-shCD73 and PC9GR-shCD73 cells indicated that CD73 silencing and EGFR-TKI treatment increased apoptosis (Fig. 4B). Annexin V/PI double staining flow cytometry assay revealed that the apoptosis rates were significantly increased in a dose-dependent manner in PC9ER-shCD73 and PC9GR-shCD73 cells treated with EGFR-TKI, but not in NC and untreated cells (Fig. 4D). Collectively, these results suggest that the addition of CD73 inhibition to EGFR-TKI can exert antitumor effects through cell cycle arrest and increase apoptosis in EGFR-TKI–resistant NSCLC.
Discussion
This study demonstrated that CD73 overexpression was significantly associated with shortened survival in EGFR-mutant NSCLC, and inhibition of CD73 overcame resistance to first-generation EGFR-TKI in EGFR-mutant lung cancer cell lines.
The EGFR signaling pathway in cancers has been shown to have a close relationship with CD73 in several studies [17,18,20]. A previous study has reported increased expression of CD73 in EGFR-mutant NSCLC [23]. Although the exact mechanism causing CD73 overexpression in EGFR-mutant NSCLC has not been fully elucidated, a recent study showed that CD73 expression is regulated through the EGFR-ERK signaling pathway, which is consistent with our study [24].
In this study, CD73 overexpression in NSCLC was associated with shorter survival, particularly in EGFR-mutant patients. Previous studies reporting similar results assessed the relationship between CD73 expression and prognosis mainly in resectable stage lung cancer [18,23]; however, this study is the first to report that CD73 overexpression was associated with shorter survival in patients in a metastatic setting.
We demonstrated that CD73 expression changed before and after EGFR-TKI treatment, and the pattern of changes was not related to an acquired EGFR T790M mutation as a resistance mechanism. A recent study reported that CD73 expression after EGFR-TKI treatment was significantly increased compared with that at baseline in PD-L1 strongly positive tumors [25]. These results indicate that the tumors of some patients who have acquired resistance to EGFR-TKI might evolve in the direction of immune evasion through the adenosine pathway.
In addition to the immunosuppressive effect of the adenosine pathway, there is increasing evidence that CD73 directly contributes to tumor progression [13,17–20]. Previous studies have reported a key role of CD73 in epithelial-to-mesenchymal transition, angiogenesis, and stemness [19,26,27].
Here, we established EGFR-TKI–resistant cell lines transfected with shRNA against CD73 to evaluate the direct effect of CD73 on cancer cells. CD73 silencing had no inhibitory effects on the growth of most cells. Thus, we assessed the antitumor effect of combining CD73 inhibition with EGFR-TKI treatment. EGFR-TKI synergistically inhibited cell growth in PC9ER-shCD73 and PC9GR-shCD73 cells compared to NC cells. Using a colony-forming assay, we confirmed the long-term effect of the combination of CD73 silencing and first-generation EGFR-TKI in resistant cells. These in vitro results support data from a recent phase Ib/2 study of a combination of CD73 inhibitor and osimertinib, showing prolonged PFS in patients with EGFR-mutant T790M-negative NSCLC [28].
We revealed that the combination of CD73 silencing and EGFR-TKI inhibited cell growth through G0/G1 phase cell cycle arrest and promoted apoptosis. This cell cycle arrest can be explained by the increased p21 and decreased cyclin D1 levels. These findings are consistent with those of previous studies [13,29,30].
In our in vitro study, we demonstrated that CD73 inhibition affected sensitivity to EGFR-TKI in EGFR-TKI–resistant cell lines, but not in EGFR-TKI–sensitive cell lines. However, our research cannot explain whether and how CD73 contributes to the development of resistance to EGFR-TKI. Another limitation of this study is that the role of CD73 in resistance to third-generation EGFR-TKIs was not shown. Although some studies have reported that CD73 is involved in drug resistance in other cancers [31], this is the first study to report the possible role of the adenosine pathway in resistance to first-generation EGFR-TKIs in NSCLC.
In conclusion, CD73, which is often overexpressed in lung cancers, is a poor prognostic factor, especially in patients with EGFR-mutant NSCLC. We demonstrated that inhibition of CD73 overcame resistance to first-generation EGFR-TKI in EGFR-mutant lung cancer cell lines by inducing cell cycle arrest and apoptosis. Further studies exploring the precise role of the adenosine pathway in the mechanism of resistance to EGFR-TKI are warranted to incorporate new drugs targeting CD73 for the treatment of patients with EGFR-TKI-resistant NSCLC.