Introduction
Lung cancer has become a remarkable example of precision medicine among solid tumors, because of the progress in understanding genomic landscape and the development of targeted therapies [1]. Epidermal growth factor receptor (EGFR) mutations are prevalent oncogenic drivers in lung adenocarcinoma that confer susceptibility to tyrosine kinase inhibitors (TKIs) [2–4]. EGFR exon 19 deletion and the L858R mutation are the most frequent sensitizing mutations in EGFR [5].
EGFR kinase domain duplication (EGFR-KDD) is a rare mutation that represents 0.2% of EGFR-mutated lung adenocarcinoma [6]. In contrast to ligand-dependent EGFR activation in typical conditions, EGFR-KDD forms ligand-independent intra-molecular dimers and amplifies downstream signal with ligand-dependent inter-molecular dimers and multimers [7–9]. Notably, discovery of EGFR-KDD as a novel oncogenic driver has led to few functional studies [8,10] and acquired resistance mechanism of EGFR-KDD against EGFR TKIs is not fully understood.
The EGFR T790M mutation is the most frequent driver of acquired resistance in approximately 60% of patients with sensitizing EGFR mutations who experience progression after treatment with 1st and 2nd generation EGFR TKIs [11,12]. Other mechanisms involve on-target mutations, bypass signaling cascades, and histological transformation [13].
In this study, we established patient-derived cell lines and developed EGFR-KDDT790M Ba/F3 models to characterize and evaluate drug sensitivity of T790M-positive EGFR-KDD mutated cells. The T790M-positive EGFR-KDD mutation were identified from a patient whose tumor acquired the T790M mutation as a resistance mechanism to erlotinib. We also suggest that the EGFR C797S mutation, in combination with the EGFR-KDDT790M mutation, mediates a potential resistance mechanism to osimertinib, based on N-ethyl-N-nitrosourea (ENU) mutagenesis screening results.
Materials and Methods
1. Targeted next-generation sequencing and fluorescence in situ hybridization
The DNA extracted from formalin-fixed paraffin-embedded tumor tissues and patient malignant pleural effusion were sequenced using an next-generation sequencing (NGS) panel named Seoul National University Hospital (SNUH) FIRST Lung Cancer Panel (LCP) [14]. The sequencing coverage and quality statistics for each sample are summarized in S1 Table.
EGFR fluorescence in situ hybridization (FISH) was performed as previously described [15]. We calculated the EGFR copy number with an LSI EGFR Spectrum Orange/CEP7 Spectrum Green probe kit (Abbott Molecular) and performed analysis using Colorado scoring criteria. FISH probe signals were detected with the Olympus BX51TRF microscope (Olympus, Tokyo, Japan).
2. Patient-derived cell line
The SNU-4784 cell line was derived from a malignant pleural effusion in a patient with pulmonary adenocarcinoma who was diagnosed with the EGFR-KDD mutation by targeted NGS. The SNU-4784 cell line was provided and authenticated with short tandem repeat profiling by the Korean Cell Line Bank (KCLB, Seoul, Korea) (S2 Table). The cells were grown in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 2 mM L-glutamine.
3. Droplet digital polymerase chain reaction
Genomic DNA from the patient’s malignant pleural effusion was isolated using Exgene Cell SV mini kit (GeneAll, Lisbon, Portugal). DNA fragments were packaged into droplets using a QX200 droplet generator (Bio-Rad, Hercules, CA) and Thr 790 sites were amplified with specific probes. Droplets with amplified DNA fragments were analyzed using a QX200 droplet reader (Bio-Rad) according to the manufacturer’s guidelines. Data and graphs were created with QuantaSoft Analysis Pro software.
4. Cell lines and reagents
The 293FT (ATCC PTA-5077, discontinued) cell line was purchased from ATCC (Manassas, VA). The Ba/F3 cell line was purchased from DSMZ (Braunschweig, Germany). Ba/F3 cells were grown in RPMI-1640 media supplemented with 10% FBS, 1% penicillin/streptomycin, 2 mM L-glutamine, and 4 ng/mL interleukin (IL)-3 (ProSpec, Rehovot, Israel). The 293FT cells were grown in DMEM supplemented with 10% FBS and 2 mM L-glutamine. Gefitinib, erlotinib, afatinib, dacomitinib, osimertinib, and lazertinib were purchased from Selleck Chemicals (Boston, MA).
5. Cloning and construction of EGFR-KDD Ba/F3 cell lines
The pBabe EGFRWT was kindly provided by Matthew Meyerson (Dana-Farber Cancer Research Institute, Boston, MA; Addgene plasmids #11011). The pBabe EGFRT790M was purchased from Addgene (Addgene, Watertown, MA; Addgene plasmid #32070). The pBabe EGFRC797S was generated by site-directed mutagenesis (Agilent Technologies, Santa Clara, CA) of the pBabe EGFRWT [16]. The EGFR-KDDWT (pMSCV EGFR-KDD) vector was kindly provided by Dr. Christine M. Lovly (Vanderbilt-Ingram Cancer Center, Nashville, TN). The pBabe EGFR-KDDD1T, EGFR-KDDD2T, EGFR-KDDBDT, EGFR-KDDT/T+C were generated with an In-Fusion HD Cloning Kit (Takara Bio USA, Inc., Mountain View, CA) and specific primers (S3 Table). Each of EGFR-KDD constructs were transduced into Ba/F3 cells and cells were selected with puromycin (2–16 μg/mL).
6. PCR and sequencing
Genomic DNA and mRNA were isolated from the SNU-4784 cell line using ALL-prep DNA/RNA micro kit (Qiagen, Venlo, Netherlands). The DNA of the EGFR-KDD Ba/F3 cell lines was isolated using an Exgene Cell SV mini kit (GeneAll, Lisbon, Portugal). For use in cloning, the EGFR exon 18–25 of pBabe EGFRWT, pBabe EGFRT790M, and pBabe EGFRC797S were amplified with specific primers (S3 Table) using CloneAmp HiFi PCR Premix (Takara Bio USA, Inc.). DNA sequences were analyzed by Sanger sequencing.
7. Cell viability assay
The EGFR-KDD Ba/F3 cell lines were cultured in 96-well plates with EGFR TKIs in RPMI-1640 media for 72 hours. EGFR TKIs were diluted from 10 mM to 0.01 nM. Cell viability was measured using Chromo-CK (Chromogen, Korea) and absorbance at 450 nm was detected using microplate reader (BioTek, Winooski, VT). SNU-4784 cells were cultured as described above. Cell viability was analyzed using a Cell Titer Glo-Luminescent cell viability assay (Promega, Madison, WI). We measured luminescent signal with a PerkinElmer Victor Light 1420 Luminescence Counter (PerkinElmer, Waltham, MA). The IC50 values were calculated with Sigmaplot 10.0 software (Systat Software Inc., San Jose, CA) and the graphs were created using GraphPad Prism 7 software (GraphPad Software, San Diego, CA). Experiments were independently repeated three times.
8. Immunoblot assay
The EGFR-KDD Ba/F3 cell lines and SNU-4784 cells were cultured on 6-well plates and treated with EGFR TKIs for 4 hours. Cells were lysed with 10× cell lysis buffer (Cell Signaling Technology, Danvers, MA), phenylmethylsulfonyl fluoride (Sigma, St. Louis, MO), PhosSTOP (Roche, Basel, Switzerland), and a proteinase inhibitor cocktail (Merck, Kenilworth, NJ). Protein was quantified using a protein assay dye reagent concentrate (Bio-Rad). Samples were loaded and separated using NuPAGE Bis-Tris Gels (Invitrogen). Separated samples were transferred to polyvinylidene difluoride membranes (Bio-Rad), and signal was detected with ECL Prime Western Blotting Detection Reagent (GE Healthcare, Chicago, IL). Total EGFR (#4267s), phospho-EGFR (#3777s), total Akt (#4685), phospho-Akt (#4060s), total Erk p42/p44 (#9102), phospho-Erk (#9106), and GAPDH (#5174) antibodies were purchased from Cell Signaling Technology. Experiments were independently repeated three times.
9. ENU mutagenesis screening
The EGFR-KDDBDT Ba/F3 cells were seeded at 5×106 cells/ml in 6-well plates and exposed to 50 μg/mL ENU for 24 hours. After that, cells were washed with RPMI-1640 media three times and then were cultured for 3 days. The cultured cells were seeded in 96-well plates with 2 μM osimertinib in RPMI-1640. We observed cells by light microscopy and periodically changed the osimertinib-containing media. DNA extracts from the drug-resistant clone were amplified with PCR and analyzed by Sanger sequencing.
Results
1. Patient and clinical history
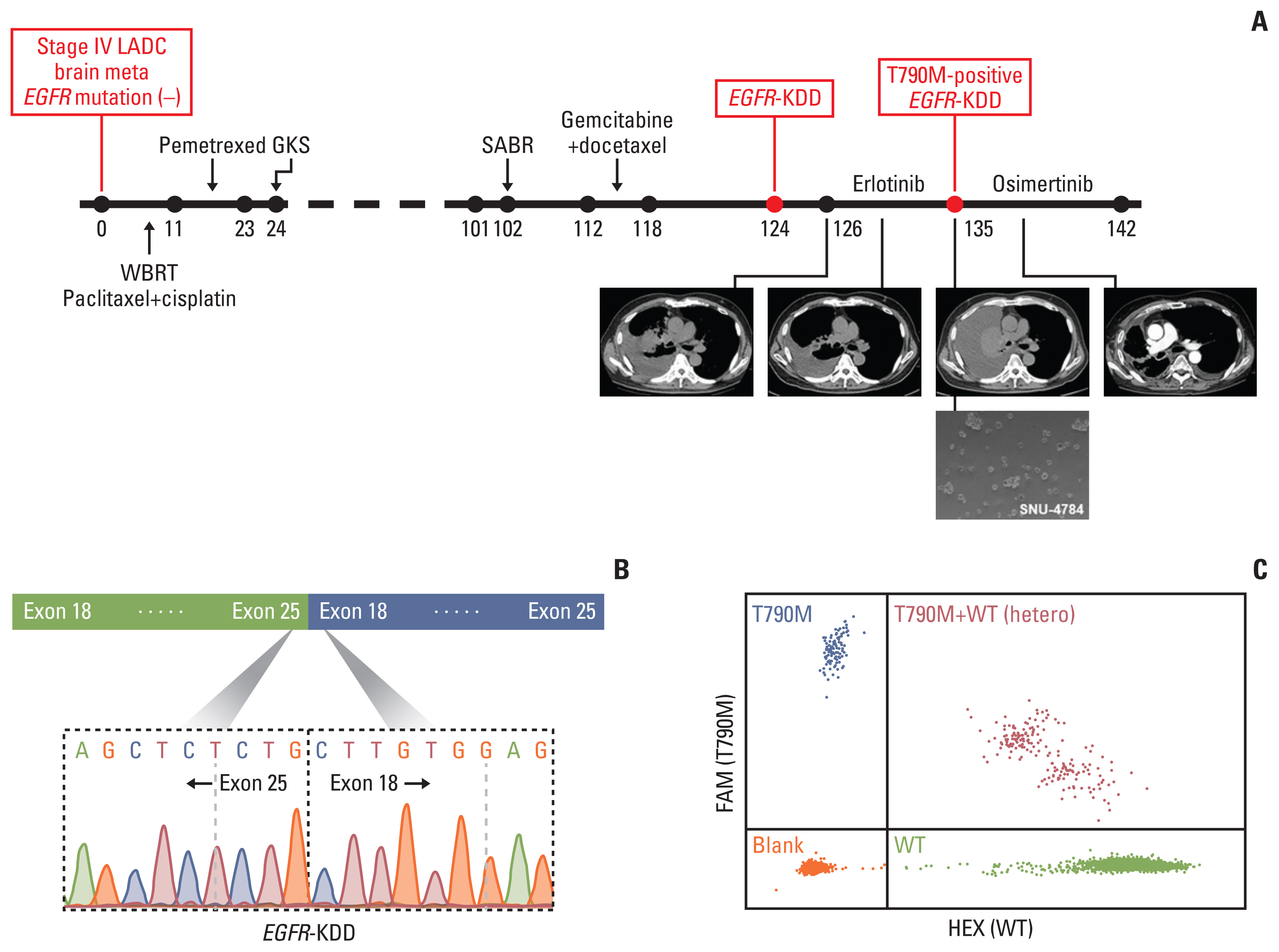
A 56-year-old man, who had never smoked, was diagnosed with stage IV lung adenocarcinoma with metastasis to the brain. After his disease progressed following third-line chemotherapy, tumor tissue was newly obtained from the primary lung tumor. NGS-based multigene panel testing detected the EGFR-KDD mutation, whereas other common EGFR mutations were absent (S4A Fig.). The presence of EGFR-KDD was confirmed with further DNA sequencing (Fig. 1B). Based on this finding, erlotinib treatment was initiated and the patient achieved a partial response. After 8 months of erlotinib therapy, the cancer progressed with increased pleural effusion, bone metastases, and peritoneal seeding. Upon disease progression, malignant cells derived from pleural effusion were collected for genetic profiling. Then, both EGFR-KDD and the EGFR T790M mutation were detected in erlotinib-resistant tumor cells and confirmed with droplet digital polymerase chain reaction (Fig. 1C, S4B and S5 Figs.). However, we could not confirm whether T790M occurred in one domain or both of EGFR-KDD. FISH analysis showed no evidence of EGFR amplification (S6 Fig.). The patient received osimertinib and achieved a partial response for 7 months. Clinical history is summarized in Fig. 1A.
2. Characteristics of the patient-derived cell line
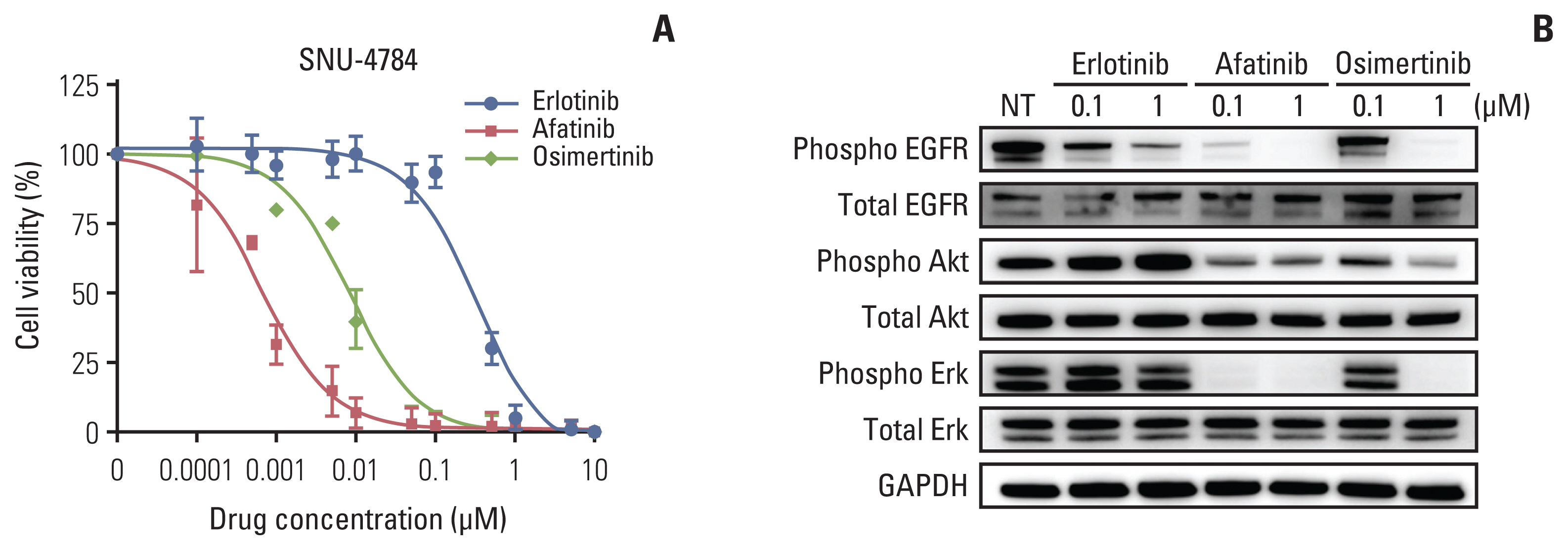
We confirmed the EGFR-KDD mutation in the patient-derived cell line SNU-4784 by Sanger sequencing; unexpectedly, the T790M mutation was not found in this cell line (S7A and S7B Fig.). We performed cell viability and immunoblot assays to determine the efficacy of EGFR TKIs in SNU-4784 cell line. We found that afatinib (IC50, 0.70±0.16nM) and osimertinib (IC50, 8.98±1.71 nM) inhibited cell viability (Fig. 2A, S8 Table). Furthermore, afatinib inhibited expression of phospo-EGFR, phospho-Akt, and phospho-Erk signals that are driven by activated EGFR (Fig. 2B) [17]. These results indicate that the EGFR-KDD mutation is sensitive to EGFR TKIs, especially afatinib, and this was demonstrated in a previous study [8].
3. Characteristics of T790M-positive EGFR-KDD Ba/F3 models
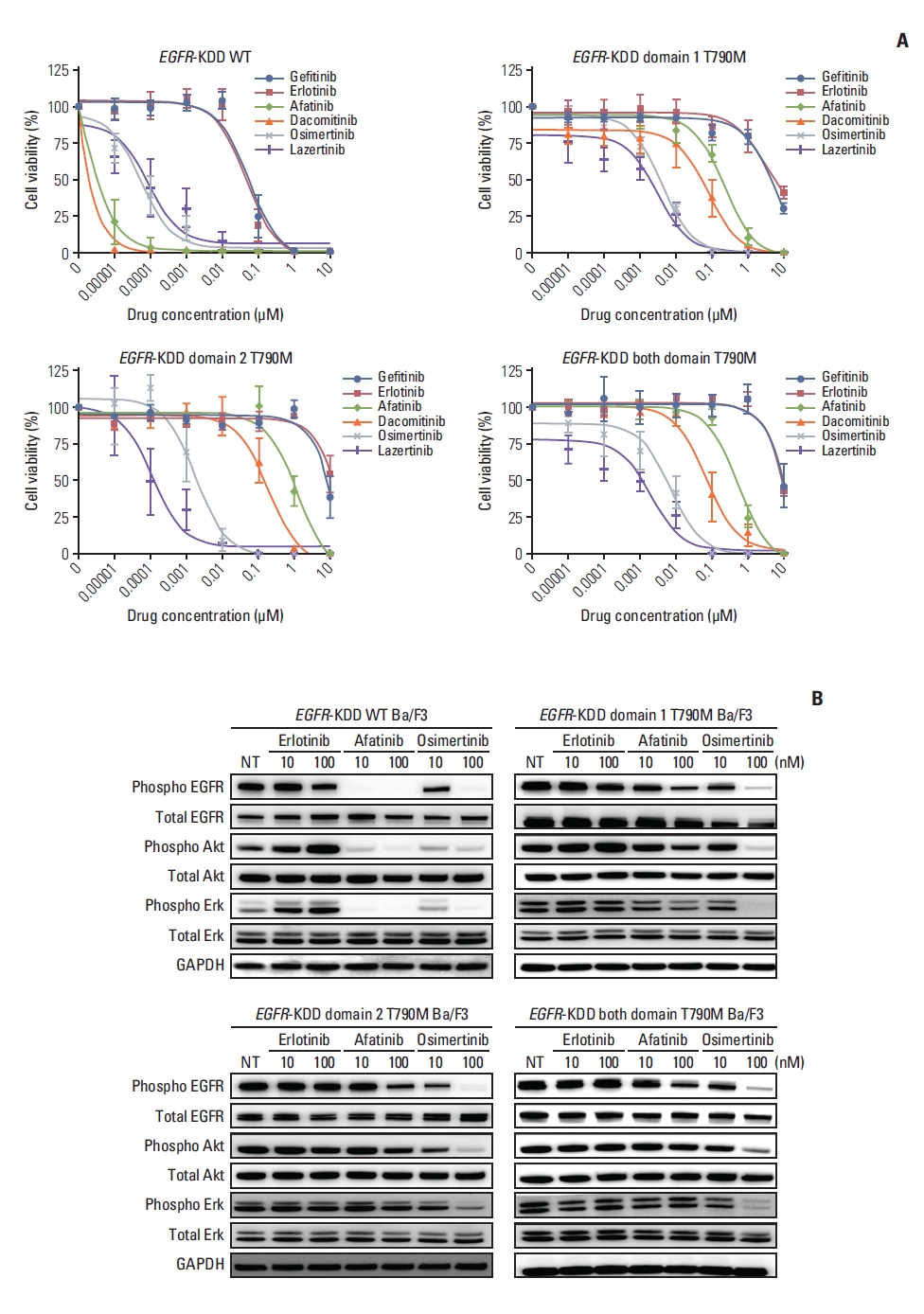
To characterize the T790M-positive EGFR-KDD mutation, we established several EGFR-KDDT790M Ba/F3 models. These included EGFR-KDD domain 1 T790M (EGFR-KDDD1T), EGFR-KDD domain 2 T790M (EGFR-KDDD2T), and EGFR-KDD both domain T790M (EGFR-KDDBDT) (S9 Fig.), and we performed cell viability assays and immunoblot assays with these Ba/F3 cell lines. We found that the EGFR-KDDWT Ba/F3 cells were sensitive to 2nd generation EGFR TKIs afatinib (IC50, < 0.01 nM) and dacomitinib (IC50, < 0.01 nM), in addition to 3rd generation EGFR TKIs osimertinib (IC50, 0.07±0.04nM) and lazertinib (IC50, 0.21±0.2 nM) (Fig. 3A, S10 Table). These results were consistent with our findings in a SNU-4784 cell line with an identical EGFR-KDDWT mutation (Fig. 2A). However, the EGFR-KDDT790M Ba/F3 cell lines were only sensitive to 3rd generation EGFR TKIs, osimertinib and lazertinib. Notably, lazertinib, an irreversible 3rd generation EGFR TKI, strongly inhibited the EGFR-KDDT790M mutation (Fig. 3A, S10 Table) [18]. Also, osimertinib inhibited phospho-EGFR, phospho-Akt, and phospo-Erk protein expression in EGFR-KDDT790M Ba/F3 cell lines. In contrast, 1st and 2nd generation EGFR TKIs did not effectively inhibit those signals (Fig. 3B). Thus, the EGFR T790M mutation, in combination with EGFR-KDD, induced resistance against 1st and 2nd generation EGFR TKIs.
4. ENU mutagenesis screening to investigate a resistance mechanism of EGFR-KDDT790M against osimertinib
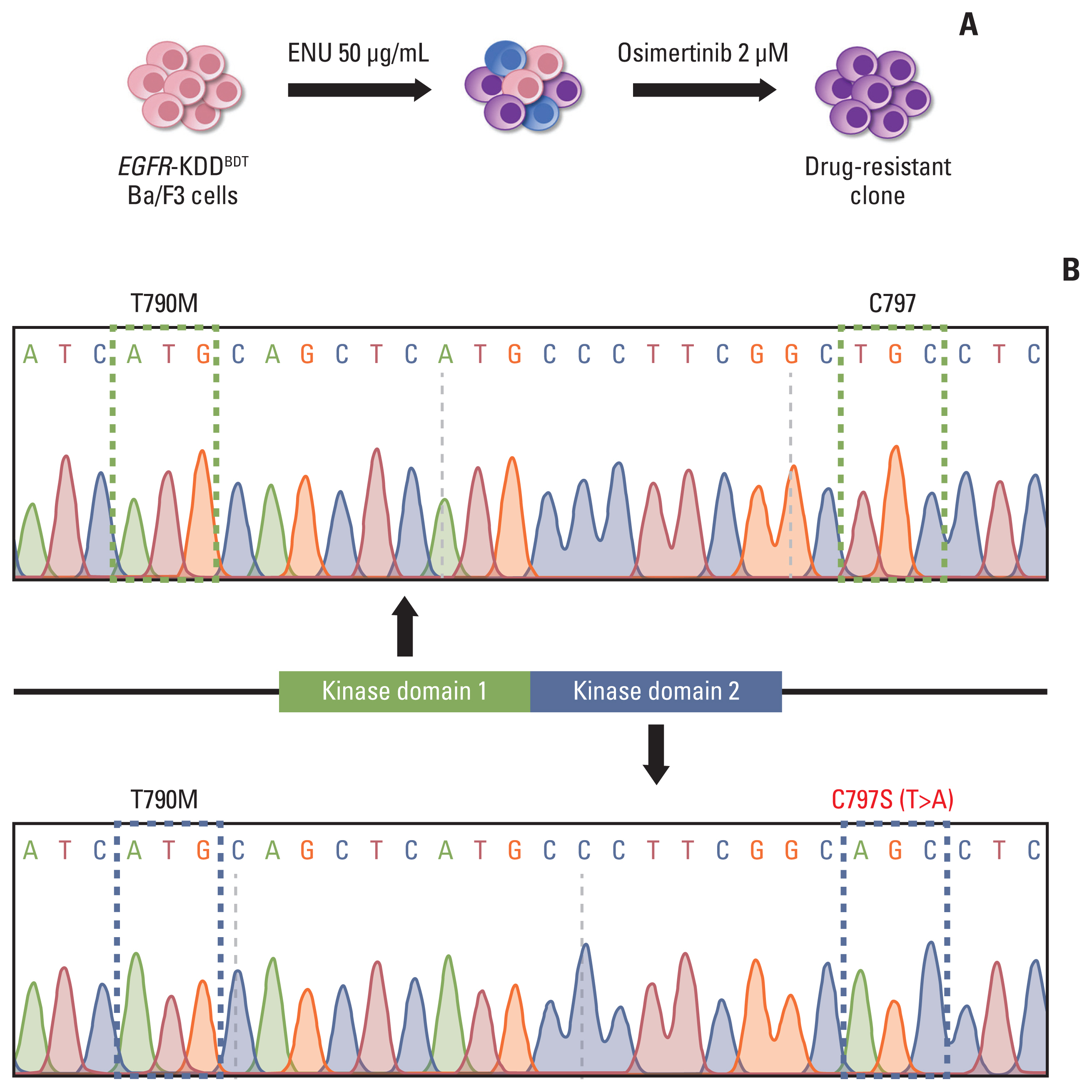
To identify the resistance mechanism against 3rd generation EGFR TKIs, we performed ENU mutagenesis screening in EGFR-KDDBDT Ba/F3 cells treated with osimertinib. We mutagenized cells via exposure to 50 μg/mL ENU and selected resistant cells with 2 μM/mL osimertinib (Fig. 4A). We sequenced DNA of the emergent resistant cells by Sanger sequencing and found the C797S mutation in kinase domain 2 of osimertinib-resistant EGFR-KDDBDT cells (Fig. 4B). Importantly, all osimertinib-resistant clones had the identical C797S mutation (Fig. 4B, S11 Fig.). These results suggest that kinase domain 2 of EGFR-KDD is more vulnerable than domain 1, and that the C797S mutation underlies a potential mechanism for EGFR-KDDT790M resistance against osimertinib.
5. The EGFR-KDDT/T+C Ba/F3 model indicating an acquired resistance mechanism
To characterize the EGFR-KDD domain 1 T790M/domain 2 cis-T790M+C797S mutation, we established an EGFR- KDDT/T+C Ba/F3 model. We performed cell viability assays and immunoblot assays to investigate the efficacy of EGFR TKIs in EGFR-KDDT/T+C Ba/F3 cells. Cells were resistant to 1st and 2nd generation EGFR TKIs, and also 3rd generation EGFR TKIs were not effective at inhibiting cell growth. (Fig. 5A). Furthermore, EGFR TKIs did not inhibit phospho-EGFR, phospho-Akt, or phospho-Erk protein expression (Fig. 5B). We then performed additional cell viability assays and immunoblot assays to identify the effect of a cetuximab combination treatment, a known therapeutic strategy for overcoming the C797S positive mutation [19,20]. Cells were exposed to EAI045 (an allosteric EGFR inhibitor) or brigatinib, in combination with cetuximab. We found that the cetuximab combination treatment did not inhibit the proliferation of the EGFR-KDDT/T+C Ba/F3 cells (Fig. 5C). In addition, the growth of EGFR-KDDT/T+C Ba/F3 cells in IL-3–free media was the most aggressive among the EGFR-KDD Ba/F3 cell lines (Fig. 5D). Taken together, our findings indicate that the cis-C797S mutation in kinase domain 2 of EGFR-KDDT790M induced drug resistance and rapid cell proliferation.
Discussion
Although EGFR-KDD was first reported in a glioblastoma, the mutation also exists in lung cancers and is a therapeutic target of EGFR TKIs [8,10,21,22]. The in-tandem and in-frame duplication of exons 18–25 encodes a permanently activated kinase with in vitro oncogenic potential [7,8].
Up to 10% of EGFR wild type patients still respond to EGFR TKIs, although the molecular basis for the response is unknown [23,24]. This gap in knowledge may be due to conventional PCR-based exon sequencing that can fail to detect rare EGFR structural variants, including EGFR-KDD and EGFR fusions, due to large intragenic repeats and/or intronic breakpoints. Thus, comprehensive molecular profiling is important to identify all genomic alterations that may lead to new therapeutic targets.
Since EGFR-KDD was discovered in a patient with metastatic lung adenocarcinoma, the duplication has been reported in more than 10 non–small cell lung cancer (NSCLC) patients, many of whom have never smoked. Patients with EGFR-KDD, including the patient in our study, are responsive to EGFR TKI. However, a recent study showed that two of the five NSCLC patients with EGFR-KDD did not respond to EGFR TKI therapies, suggesting the existence of a separate uncharacterized primary resistance mechanism [8,25].
Here we studied a patient with lung adenocarcinoma with an EGFR-KDD mutation, which was identified with NGS-based multigene panel testing. Consistent with previous reports, the patient was responsive to erlotinib as an EGFR TKI therapy [6]. Although patient-derived cell line lacks of EGFR T790M mutation (EGFR-KDDWT), that might be caused by clonal selection during establishment of patient-derived cell line due to low EGFR T790M proportion of patient’s tumor (S2 Fig.), we reaffirmed the finding of EGFR-TKI sensitivity against EGFR-KDDWT mutation with patient-derived cell line.
Due to the rarity of EGFR-KDD cases, there are few studies on the acquired resistance mechanism of EGFR-KDD tumors. The amplification of EGFR was previously demonstrated as one of acquired resistance mechanisms of EGFR-KDD tumors [8,25,26]. Also, the existence of EGFR T790M mutations in the post-TKI biopsy tumors, which might be related to developing resistant clones, was reported in previous studies [22,25].
Our results demonstrated that T790M contributed to the acquired resistance of EGFR-KDD tumors to erlotinib, which was further supported by the response to osimertinib, and this was also demonstrated with EGFR-KDDT790M Ba/F3 models. Our study provides a new report to indicate a drug can overcome the resistance mechanism in a patient with NSCLC harboring EGFR-KDD.
Additionally, our results indicated that the cis-C797S mutation underlies a resistance mechanism that acts with the EGFR-KDDT790M mutation against 3rd generation TKIs. The C797S mutation is known to cause acquired resistance to 3rd generation EGFR TKIs, and there is no targeted therapy for the cis-T790M/C797S mutation [27–29]. Cetuximab combination treatments were studied as a strategy to overcome the C797S mutation, but the EGFR-KDDT/T+C mutation was resistant to those strategies [19,20].
In summary, we report a patient with lung adenocarcinoma harboring EGFR-KDD who experienced an anti-tumor response to erlotinib, as well as acquired resistance caused by T790M. Tumor progression in the patient was inhibited by osimertinib, and these findings were supported by our in vitro studies. Also, our results suggest that the EGFR-KDDT/T+C mutation mediates resistance mechanism against 3rd generation EGFR TKIs and cetuximab combination strategies. Further studies should target this type of mutation to benefit EGFR-KDD patients who develop acquired resistance.