Introduction
Breast cancer constitutes approximately 30% of cancers in women, with a mortality-to-incidence ratio of 15% [1]. While early and middle stage breast cancer patients can undergo radical surgical resection, postoperative recurrence remains a significant challenge for clinicians [2].
Minimal residual disease (MRD) has been identified as the primary cause of breast cancer recurrence, with patients exhibiting detectable MRD experiencing shorter disease-free survival (DFS) and overall survival [3-6]. Nevertheless, the genomic features of patients with detectable MRD and the mechanisms underlying its formation warrant further investigation.
In contrast to clinical imaging, the identification of MRD can provide early indications of patient recurrence [3,5,7], thereby affording clinicians an opportunity for timely intervention. Furthermore, ongoing MRD-guided prospective intervention studies on breast cancer are currently being conducted [8-10]. However, the selection of suitable drugs for patients with detectable MRD presents a significant challenge.
The utilization of next-generation sequence (NGS) technology has exhibited significant advancements in comprehending the genome of breast cancer, rendering it a crucial instrument for the diagnosis and treatment of this disease [11-14]. Presently, the National Comprehensive Cancer Network (NCCN) guidelines endorse the use of NGS for the detection of mutations in BRCA1, BRCA2, PALB2, PIK3CA, ESR1, ERBB2, and fusions of NTRK, RET, as well as the calculation of tumor mutation burden (TMB) values in breast cancer patients, thereby facilitating the selection of targeted therapy [15].
In this study, 80 patients with breast cancer who had undergone tumor genetic testing and postoperative MRD testing with NGS-based 1,021 cancer-related gene panels were enrolled, in an attempt to characterize the genomic profile and the treatment strategies for patients with detectable MRD.
Materials and Methods
1. Patient enrollment
We screened 80 patients with breast cancer who had undergone both tumor genetic testing and postoperative MRD testing from Army Specialty Medical Center between June 2017 and January 2023. The criteria for the patient’s enrollment were as follows: (1) pathological diagnosis as breast cancer, (2) no distal metastasis, (3) radical surgery and R0 resection, (4) 1,021 cancer-related gene testing of the tumor, (5) plasma MRD testing performed within 3 months after surgery and before adjuvant therapy. Clinical information and testing information were collected for every patient.
2. Genetic testing of tumor tissue
For patients receiving neoadjuvant therapy, needle biopsies of the tumor were obtained before neoadjuvant therapy. For patients without receiving neoadjuvant therapy, tumor tissues were obtained after surgery. All tumor samples were prepared into formalin-fixed and paraffin-embedded tissues (FFPE). Five milliliters of peripheral blood was drawn from each patient using an EDTA tube as control. All samples were sent to Geneplus-Beijing Ltd. (Beijing, China) for NGS-based genetic testing of 1,021 cancer-related genes (S1 Table). Briefly, genomic DNA from FFPE and peripheral blood were extracted by Maxwell 16 FFPE Plus LEV DNA Purification kit (AS1135, Promega, Madison, WI) and CWE9600 Blood DNA kit (CW2531S, CWBiotech, Taizhou, China), respectively. Genomic DNA was broken into fragments with a peak value of 300 bp by the Covaris S2 system. Library construction was performed by NEB Next Ultra II DNA Kits (E7645, NEB, Ipswhich, MA). A customized 1,021 gene probe set was used to enrich the target region of library DNA. Finally, the captured library was sequenced on the Gene+Seq-2000 sequencer (Geneplus-Suzhou Biomedical Engineering Corporation, Suzhou, China). The sequencing depth of tumor tissue was greater than 500×, and the sequencing depth of peripheral blood cells was greater than 150×.
3. MRD testing of postoperative plasma
Twenty milliliters peripheral blood was drawn from each patient within 3 months after surgery and before adjuvant therapy by using Cell-Free DNA BCT Blood Collection Tube (218962, Streck, Omaha, NE) and sent to Geneplus-Beijing Ltd. for NGS-based MRD testing. Briefly, peripheral blood was centrifuged at 2,500 ×g for 10 minutes. The supernatant was transferred to a centrifuge tube and centrifuged at 16,000 ×g for 10 minutes to remove residual cell debris. The supernatant was plasma. Circulating free DNA (cfDNA) from plasma was extracted by The MAGMAX cell-free DNA ISO Kit (A29319, Life Technology, Carlsbad, CA). The library construction was the same as the genetic testing of tumor tissue. The sequencing depth of cfDNA was greater than 10,000×.
4. Sequencing data analysis
Raw data were filtered to remove adaptor and low-quality reads by fastp software ver. v0.23.2. The filtered reads were further aligned to the human genome hg19 by Burrows Wheel Aligner software ver. 0.6.2.
For genetic testing of tumor tissue, somatic single-nucleotide variants (SNVs) and small insertions or deletions (Indels) were analyzed by the MuTect2 algorithm. All reliable gene variants were supported by ≥ 5 high-quality sequencing reads. The somatic copy number variant (CNV) was analyzed by CONTRA ver. 2.0.8 software. A threshold of 0.75 and 1.25 was used to delineate the cutoff for CNVs loss and gain, respectively. Germline mutation was filtered by using blood cell sequencing reads.
For MRD testing of postoperative plasma, our study used tumor-informed assays to determine the status of plasma circulating tumor DNA (ctDNA) [16]. Briefly, for tumor tissue-derived variation, ≥ 2 high-quality sequencing reads are needed for tumor-specific driver gene variations, and ≥ 4 reads are needed for passenger gene variants. Meanwhile, for non-tumor tissue-derived variations, ≥ 4 high-quality sequencing reads were required for driver gene variants, and ≥ 8 reads were required for passenger gene variants. Detection of at least one of these variants in a plasma sample was defined as ctDNA positive. ctDNA positive means detectable MRD. Conversely, if no variant was detected in a plasma sample, it was deemed to ctDNA negative. ctDNA negative means undetectable MRD.
5. Genomic characteristics analysis
Mutation characteristics were analyzed by Maftools software (ver. 2.16.0) [17]. The mutational distribution of the key genes was performed by the oncoplot function. The difference in mutation frequency between the two groups was performed by the mafCompare function. The presentation of differential genes was performed by the forestPlot function and coBarplot function. The mutually exclusive or co-occurring mutations were performed by the somaticInteractions function. The oncogenic signaling pathways were analyzed by the OncogenicPathways function. The presentation of the oncogenic signaling pathway was referred to the report of Sanchez-Vega et al. [18].
Mutational signatures were conducted by the deconstructSigs software ver. 1.8.0 [19], and the aetiologies of signatures were referred to the report of Alexandrov et al. [20] and Catalogue of Somatic Mutations in Cancer (COSMIC) database [21]. The targeted therapy analysis of each patient was referenced in the OncoKB database [22].
6. Data availability
The genetic variation information generated by NGS in this study are available through The Genome Variation Map of the National Genomics Data Center, China National Center for Bioinformation (accession number: GVM000547).
7. Statistical analysis
Fisher’s exact test was used for categorical variables and the t test was used for continuous variables by SPSS software ver. 26 (IBM Corp., Armonk, NY). Kaplan-Meier curves were performed by GraphPad Prism software ver. 8.0.2 (GraphPad Software Inc., San Diego, CA). A p-value less than 0.05 indicates a difference.
Results
1. Clinical characteristics of patients
A total of 80 breast cancer patients underwent genetic testing of baseline tumor tissue and MRD testing of postoperative plasma, of which 18 patients were detectable and 62 patients were undetectable for MRD testing (S2 Table). The mean age of patients with detectable MRD was 45.56±1.69 years old, which showed no difference from patients with undetectable MRD (p=0.470). Most patients with detectable MRD were stage III (67 %, 12/18), lymph node status N3 (44%, 8/18), human epidermal growth factor receptor 2–positive (44%, 8/18), and high-risk (61.1%, 11/18). Fisher’s exact test analysis found that patients with detectable MRD were associated with the clinical stage (p=0.028). Patients with higher clinical staging were more likely to have detectable MRD. However, it was not related to tumor size, lymph node status, molecular typing, neoadjuvant therapy, and clinical risk grades (p > 0.05) (Table 1, S2 Table). Interestingly, we found that in patients who did not receive neoadjuvant, detectable MRD was associated with stage, tumor size, and lymph node status (p < 0.05), but this was not observed in patients with neoadjuvant therapy (S3 and S4 Tables).
By the way, the median follow-up time of enrolled patients was 29 months (range, 1 to 76 months), and only four patients had relapsed. Kaplan-Meier curves showed that there was no difference in DFS between patients with detectable MRD and undetectable MRD (p=0.638; hazard ratio, 1.23; 95% confidence interval, 0.11 to 13.50), and the median DFS was not reached in both groups (S5A Fig.). Subgroup analysis showed no difference in DFS in patients with detectable MRD and undetectable MRD neither in patients who received neoadjuvant therapy nor those who did not (S5B and S5C Fig.). Follow-up data of patients are not mature enough to confirm that postoperative MRD status can predict the prognosis of patients.
2. Characteristics of tumor mutations in patients with detectable MRD
The positive rates of somatic SNV/Indel mutations in patients with detectable or undetectable MRD were 100% (18/18) and 98% (61/62), respectively. The top five somatic mutational genes for patients with detectable MRD were TP53 (67%, 12/18), PIK3CA (33%, 6/18), MAP3K1 (22%, 4/18), MLL3 (22%, 4/18), FAT1 (17%, 3/18), or GATA3 (17%, 3/18) (Fig. 1A). The top five somatic mutational genes for patients with undetectable MRD were TP53 (52%, 32/62), PIK3CA (33%, 20/62), GATA3 (10%, 6/62), ARD1A (8%, 5/62), and BRAF (7%, 4/62) (Fig. 1B). Compared with patients with undetectable MRD, mutations of MAP3K1, ATM, FLT1, GNAS, POLD1, SPEN, and WWP2 were significantly enriched in patients with detectable MRD (p < 0.05) (Fig. 1C and D). Mutational mutual exclusion and co-occurrence analysis showed that TP53 was mutational exclusive with MLL3 (p < 0.01) or GATA3 (p < 0.05) and NCOR1 was mutational co-occurrence with MAP3K1 or MLL3 (p < 0.05) in patients with detectable MRD (S6A Fig.). In patients with undetectable MRD, ARID2 was mutational co-occurrence with NSD1 (p < 0.01) and TP53 was mutational exclusive with MED12 (p < 0.05) (S6B Fig.). These results suggest that the genomic characteristics of patients with detectable MRD are different from those with undetectable MRD.
3. Oncogenic signaling pathways affected by somatic mutations
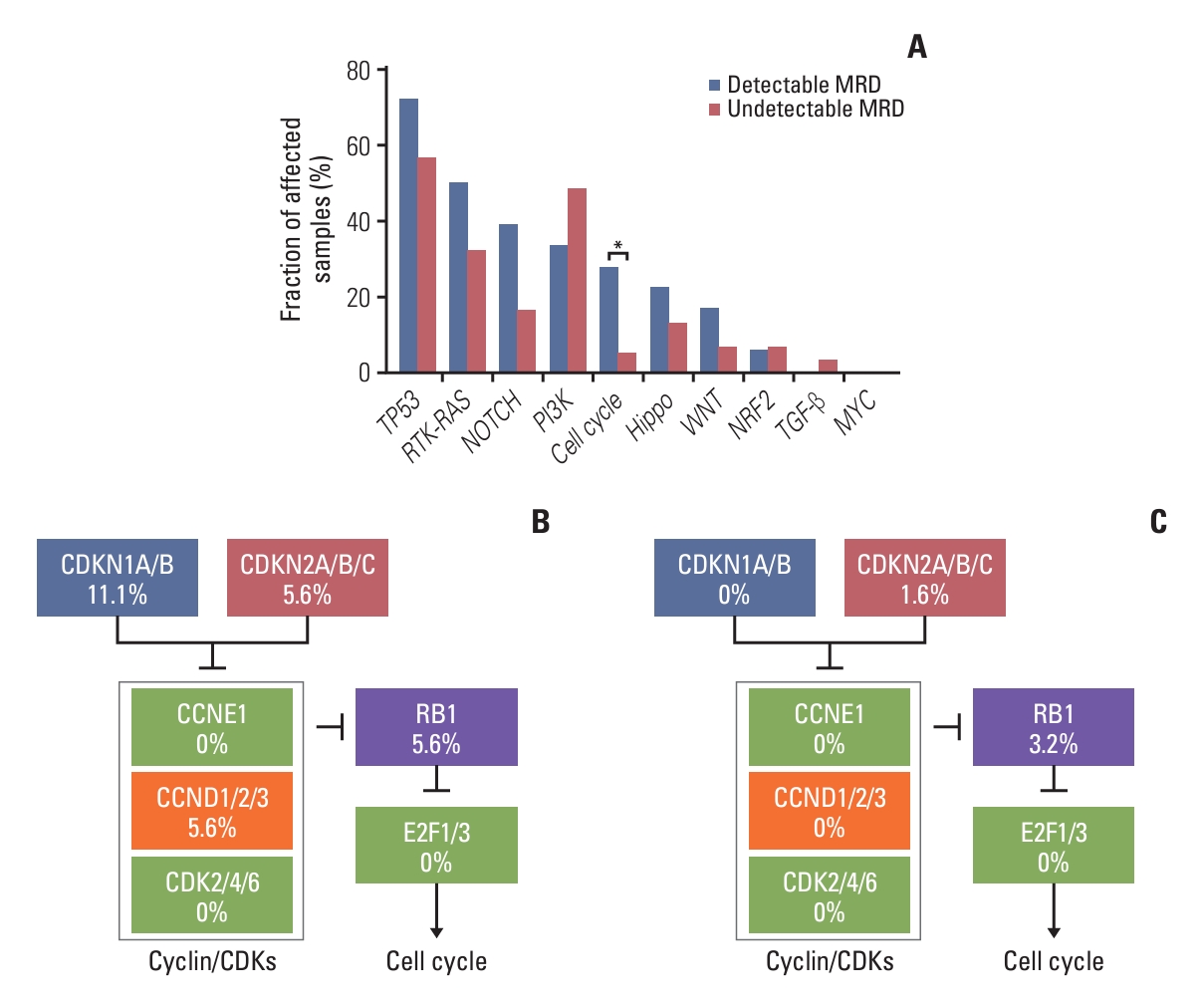
Oncogenic signaling pathways have 10 cancers related signaling pathways, such as RTK-RAS pathway, NRF2 pathway, phosphoinositide 3-kinase pathway, TP53 pathway, WNT pathway, MYC pathway, transforming growth factor β pathway, Hippo pathway, NOTCH pathway, cell cycle pathway. We found that the TP53 pathway (72.2% vs. 56.5%), RTK-RAS pathway (50.0% vs. 32.3%), NOTCH pathway (38.9% vs. 16.1%), cell cycle pathway (27.8% vs. 4.8%), Hippo pathway (22.2% vs. 12.9%), WNT pathway (16.7% vs. 6.5%) were more altered in patients with detectable MRD than those with undetectable MRD (Fig. 2A). Furthermore, alteration in cell cycle pathway was significantly different between these two groups (p=0.012) (Fig. 2A). Gene mutations of cell cycle pathway in patients with detectable MRD mainly occurred in CDKN1A/B (11.1%, 2/18), CDKN2A/B/C (5.6%, 1/18), CCND1/2/3 (5.6%, 1/18), and RB1 (5.6%, 1/18) (Fig. 2B). But, gene alteration of cell cycle pathway in patients with undetectable MRD only occurred in CDKN1A/B (1.6%, 1/62) and RB1 (3.2%, 2/62) (Fig. 2C). These results suggested that mutations in cell cycle pathway are associated with detectable MRD.
4. Mutational signatures of patients with detectable MRD
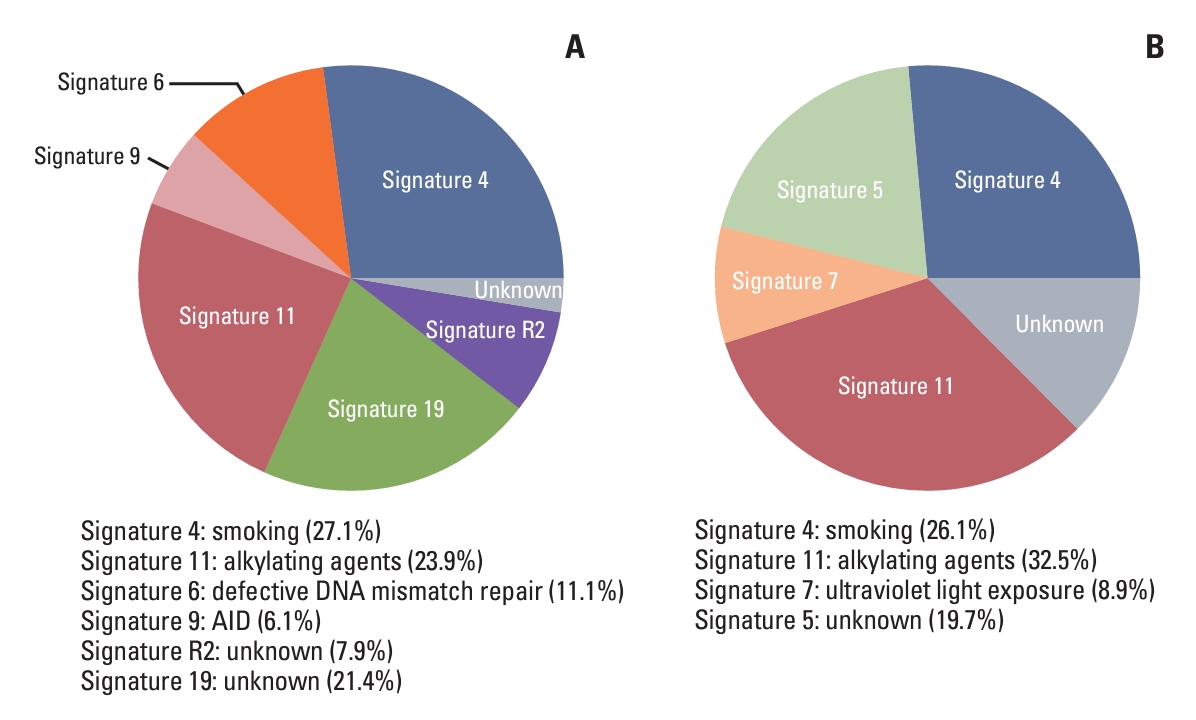
A total of 6 signatures were found in patients with detectable MRD. They are signature 4 (27.1%), signature 11 (23.9%), signature 19 (21.4%), signature 6 (11.1%), signature R2 (7.9 %), and signature 9 (6.1%) (Fig. 3A, S7A Fig.). Four signatures were found in patients with undetectable MRD, which were signature 11 (32.5%), signature 4 (26.4%), signature 5 (19.7%), and signature 7 (8.9%) (Fig. 3B, S7B Fig.). Signature 4 and signature 11 were present in both groups. The aetiologies of signature 4 and signature 11 are smoking and alkylating agents, respectively. Signature 19, signature 6, signature R2, and signature 9 were found only in patients with detectable MRD. It is noteworthy that the aetiologies of signature 6 and signature 9 are defective of DNA mismatch repair and AID-mediated somatic hypermutation (SHM), respectively (Fig. 3A). While the aetiology of signature R2 and signature 19 are unknown. Signature 5 and signature 7 were present only in patients with undetectable MRD and the aetiology of them are unknown and ultraviolet light exposure, respectively (Fig. 3B). Based on these results, we speculated that defective of DNA mismatch repair and AID-mediated SHM may be the underlying causes of detectable MRD.
5. Somatic CNV analysis
CNV gain occurred in 14 patients (77.8%, 14/18) with detectable MRD, and two of them had CNV loss. While CNV gain occurred in 42 patients (66.7%, 42/62) with undetectable MRD, including two patients with CNV loss. Compared with patients with undetectable MRD, CNV gain of CDKN2A, HOXB13, PPM1D, MPL, and VHL was significantly enriched in patients with detectable MRD (S8 Table).
6. Clinical actionability for targeted therapy
Is there an opportunity for targeted therapy in patients with detectable MRD? By using the OncoKB database (Fig. 4A), we found that 77.8% (14/18) of patients with detectable MRD had U.S. Food and Drug Administration (FDA)–approved actionable genetic markers and FDA-approved targeted drugs (level 1 therapeutic) and 88.9% (16/18) of patients had actionable genetic markers for targeted therapy (Fig. 4B). Only 62.9% (39/62) of patients with undetectable MRD could access level 1 targeted therapy (Fig. 4C, S9 Fig.). Among patients with detectable MRD, 38.9% (7/18) of patients with ERBB2 amplification had opportunities for anti–human epidermal growth factor receptor 2 therapy and 33.3% (6/18) of patients with activated PIK3CA mutation, including two patients with p.E542K, one patient with p.E545K, two patients with p.H1047R, one patient with p.H1047L (Fig. 4D, S10 Fig.), could benefit from alpelisib plus fulvestrant. There were two patients (2/18, 11.1%) with TMB-High (≥ 10 mutants/Mb) who could access pembrolizumab and one patient (1/18, 5.6%) with germline BRCA1 p.N112Ifs*7 mutation who had a chance for olaparib in patients with detectable MRD (Fig. 4D). These results suggest that the vast majority of patients with detectable MRD have an opportunity for targeted therapy.
Discussion
Previous studies have emphatically confirmed the clinical value of MRD. It is an indisputable fact that patients with detectable MRD have a poor prognosis [3-6]. But the mechanism of MRD formation remains unclear. For the first time, we attempted to interpret the mechanism of MRD formation from clinical features and baseline tumor genome.
According to the results of this study, patients with higher clinical stages were more likely to develop detectable MRD. Furthermore, patients with detectable MRD have unique genomic characteristics. We found that somatic mutations and amplification of some genes were significantly enriched in patients with detectable MRD. Meanwhile, patients with detectable MRD had specific mutational exclusion and co-occurrence characteristics.
We also found that cell cycle pathway alterations were significantly associated with detectable MRD. In the IMvigor010 study [23], tumor transcriptome from urothelial carcinoma patients showed that genes in cell cycle pathways were significantly enriched in patients with detectable MRD. This report coincides with our conclusion. These results suggest that the abnormality of genes related to the cell cycle pathway is related to the formation of detectable MRD.
In our study, signature 4 and signature 11 accounted for a high proportion both in patients with detectable and undetectable MRD. The aetiologies of signature 4 and signature 11 are smoking and alkylating agents, respectively. Studies have confirmed that smoking and alkylating agents are associated with breast cancer [24,25]. Moreover, we found that signature 6 and signature 9 were present only in patients with detectable MRD. The mutations in signature 6 are characterized by a high frequency of small insertions and deletions (less than 3 bp) associated with the etiology of DNA mismatch repair defects [21]. Defective DNA mismatch repair is most common in colorectal cancer and urothelial carcinoma, less than 3% in other tumors, and only 1%-2% in breast cancer [26]. The mismatch repair system is the main path to maintain the stability of the genome. The defects of the mismatch repair system could cause genome hypermutation and microsatellite instability [27], which could make the tumor more heterogeneous, invasive, and evade immune monitoring [28]. The etiology of signature 9 is involved in AID-mediated SHM. The main function of AID is to induce point mutations, which can lead to increased mutation load, the disorder of genome integrity, and immune escape [29]. These characteristics of mismatch repair defects and AID disorder may facilitate the formation of postoperative MRD.
Now there are more actionable mutations and corresponding targeted drugs in breast cancer [15], which greatly improve the prognosis of patients with breast cancer. Our analysis by using the OncoKB database found that 77.8% of patients with detectable MRD had a chance of level 1 targeted therapy. With the development of MRD-guided intervention studies, there is hope that patients with detectable MRD may receive targeted therapy before clinical relapse in the future. These results provide new ideas for the accurate diagnosis and treatment of breast cancer.
At the same time, this study also has some limitations. Due to the short clinical follow-up time and the small number of patients with recurrence, the postoperative MRD status is not enough to predict the prognosis of patients. This also requires us to continue to follow up with patients and improve these results in the future. The correlation between detectable MRD and clinical features mentioned in the article, as well as the significant enrichment of some somatic variations and signaling pathway variation in patients with detectable MRD, may require further data verification. The preliminary mechanisms mentioned in the article that may be related to the detectable MRD may also need to be validated by cytological and zoological models. Finally, the targeted drugs mentioned in this study are mainly used for advanced patients at present, and the evidence for the use of these targeted drugs in patients with persistently detectable MRD is insufficient.
We studied the genomic characteristics of patients with detectable MRD and found that cell cycle pathway, DNA mismatch repair defects, and AID-mediated SHM were associated with detectable MRD. We also found that the vast majority of patients with detectable MRD had the opportunity of FDA-recognized targeted therapy.