Introduction
Lymphomas are a heterogeneous group of tumors that have striking underlying genetic diversity and spatial tumor heterogeneity [1]. Pathological morphology analysis is the gold standard for lymphoma diagnosis. It is also used for lymphoma subtyping to select treatment options and predict disease prognosis. However, pathologic analysis is unable to distinguish different genetic subtypes of certain lymphomas, such as diffuse large B cell lymphoma (DLBCL). Different genetic subtypes of lymphomas have distinct clinical outcomes [2] and novel options for targeted therapies [2,3]. Therefore, it is critical to obtain genetic information in addition to histologic information in lymphomas. Genetic changes in lymphomas are also dynamic. Tumors have complex heterogeneity that can be gained or lost temporally or spatially in response to various endogenous and exogenous selection pressures, including therapy [4-6]. Technologies that capture this heterogeneity are needed to better understand these diseases throughout their progression and treatment.
In recent years, it has become possible to assess tumor-derived DNA through circulating tumor DNA (ctDNA) that is isolated from plasma. Genotyping this ctDNA can provide information concerning the genetic profiles of tumor tissue [7]. ctDNA has been investigated in Hodgkin lymphoma (HL) and non-HL using next-generation sequencing (NGS). These techniques provide an easy-to-use detection method of the genotypic information in lymphoma [8].
Several studies of ctDNA in DLBCL using NGS-customized gene panels have shown that the mutational landscape from ctDNA was highly consistent with that observed in tissue biopsies [7,9-11]. In addition, ctDNA baseline levels have proven to be a remarkably useful tool to evaluate tumor burden and clinical outcomes [7,9].
A mutation profile cannot replace the gold standard of pathological tissue-based diagnosis. However, the non-invasive detection of ctDNA might become a valid tool for certain clinical situations, such as inaccessible tumors and/or patient condition. In this study, we designed a customized ctDNA NGS panel for lymphoma that can be applied to various subtypes. We evaluated the ctDNA panel using mutation detection concordance of plasma samples with matched tissue samples from lymphoma patients. In addition, we applied the ctDNA panel to lymphoma patients and correlated the results with their respective clinical statuses.
Materials and Methods
1. Patients and study design
We collected and stored plasma and buffy coat samples at –70℃ after obtaining written informed consent. We recorded the following clinicopathological data from the electronic medical records: age, sex, date of diagnosis, date of follow-up, International Prognostic Index (IPI) score, Eastern Cooperative Oncology Group performance score (ECOG PS) and pathological information about the lymphoma tissue. The Ann Arbor (AA) staging system was used for stage assessment. We reviewed the results from the clinical staging workup, which included computed tomography, 18F-fluorodeoxyglucose positron emission tomography/computed tomography, and bone marrow biopsy.
2. Sample preparation and NGS
We collected blood samples from patients before treatment. Blood samples were collected in a cell-free DNA collection tube (Dxtube, Dxome, Seongnam, Korea), and plasma separation was done within 24 hours to 48 hours after blood collection. Plasma was separated from whole blood using double centrifugation (1,900 ×g for 15 minutes) and then transferred to fresh tubes. Plasma samples were stored at –80ºC until analysis. The ctDNA was extracted from 4 mL of plasma using magnetic circulating DNA Maxi Reagent (Dxome) according to the manufacturer’s instructions. Peripheral blood mononuclear cells (PBMCs) were used as a non-tumor control sample. The tumor samples at diagnosis for NGS were obtained from archived formalin-fixed paraffin-embedded (FFPE) tissue blocks. The tumor fractions in FFPE samples were over 80% in most cases (15/18). Genomic DNA (gDNA) was isolated from PBMC and FFPE tumor samples using the QIAamp DNA Mini Kit (Qiagen, Venlo, The Netherlands) and fragmented to a mean size of 250 base pairs using the sonicator (Diagnode, Lausanne, Switzerland). The DNA yield and size distribution were analyzed using the TapeStation 4150 (Agilent Technologies, Santa Clara, CA) and a Qubit 4.0 fluorometer (Thermo Fisher Scientific, Waltham, MA).
Library preparation was performed using 2.5-30 ng of ctDNA and 110-200 ng of sheared gDNA using the DxSeq Library prep reagent (Dxome). For each sample, PBMCs were sequenced as germline-matched controls using an identical panel and library kits targeting an average depth of > 2,500×. Target capture enrichment was done using a custom panel targeting 112 genes related to various lymphoma subtypes (S1 Table). Pooled libraries were paired-end sequenced (2×150 bp) on the NovaSeq 6000 System (Illumina, San Diego, CA). Single nucleotide variants (SNVs) and insertions/deletions (indels) were called using the PiSeq algorithm (Dxome) to differentiate low-frequency mutations from amplification artifacts and sequencing errors by calculating the genomic positions of mapped reads [12]. The analytical sensitivity of 0.24% was assumed for SNVs [12]. Copy number alteration (CNA) was called using ExomeDepth and our custom tool [13]. Variants were annotated using DxSeq software (Dxome) with public database information. We visually confirmed the identified variants using the Integrative Genome Viewer (Broad Institute, Cambridge, MA).
3. Statistical analysis
All statistical analyses were performed using Microsoft Excel (Microsoft Corporation, Redmond, WA) and MedCalc ver. 18.2.1 (MedCalc Software, Mariakerke, Belgium). Correlations between the results were evaluated using the Passing-Bablok model and Pearson’s correlation coefficient (R). Descriptive statistics are presented as proportions and medians. The intergroup comparisons for categorical variables were assessed using the Mann-Whitney test. The Kruskal-Wallis test, Fisher’s exact test, and Tukey-Kramer test were used to compare the median differences between groups. We analyzed the association between ctDNA concentration and ctDNA mutation detection using receiver operating characteristic (ROC) curve analysis. Progression-free survival (PFS) was defined as the time from diagnosis to the first relapse or death. Overall survival (OS) was measured from the date of confirmed diagnosis to the date of death for any reason or the last follow-up. Statistical significance was defined as a p-value < 0.05.
Results
1. Patients characteristics
A total of 42 patients were included in this study (Table 1). The median patient age was 60 years (range, 15 to 87 years), and 25 patients (59.5%) were men. The patients were diagnosed with the following lymphoma subtypes: (1) DLBCL (n=21), (2) follicular lymphoma (FL; n=12), (3) HL (n=5), and (4) others including marginal zone lymphoma (n=1), angioimmunoblastic T-cell lymphoma (n=1), extranodal natural killer (NK)-T cell lymphoma (n=1) and mycosis fungoides (n=1). According to the AA staging criteria, 27 patients (64.3%) had advanced stages, defined by stage III/IV. The majority of patients (85.7%) had ECOG PS of 0 to 1. Most of the patients (83.3%) were treated with chemotherapy after diagnosis. Six patients had regular disease monitoring without chemotherapy. One patient was transferred to another hospital; therefore, the specific treatment that this patient received was uncertain.
2. Concordance between tissue and liquid biopsies
NGS analysis using matched tissue samples were available for 18 of the 42 liquid biopsies (18/42, 42.9%) (S2 Table). Three hundred four and 183 SNVs or indels were identified in 27 plasma and 16 tissue, respectively, and no CNA was detected in plasma and tissue. Of all evaluated tissue biopsy specimens, 57.1% (24/42) of the samples had insufficient residual tissue amounts for NGS analysis. Eighteen tissue samples were obtained from eight lymph nodes, three bone marrow samples, three tonsils, two nasal mucosa samples, and two soft tissue samples.
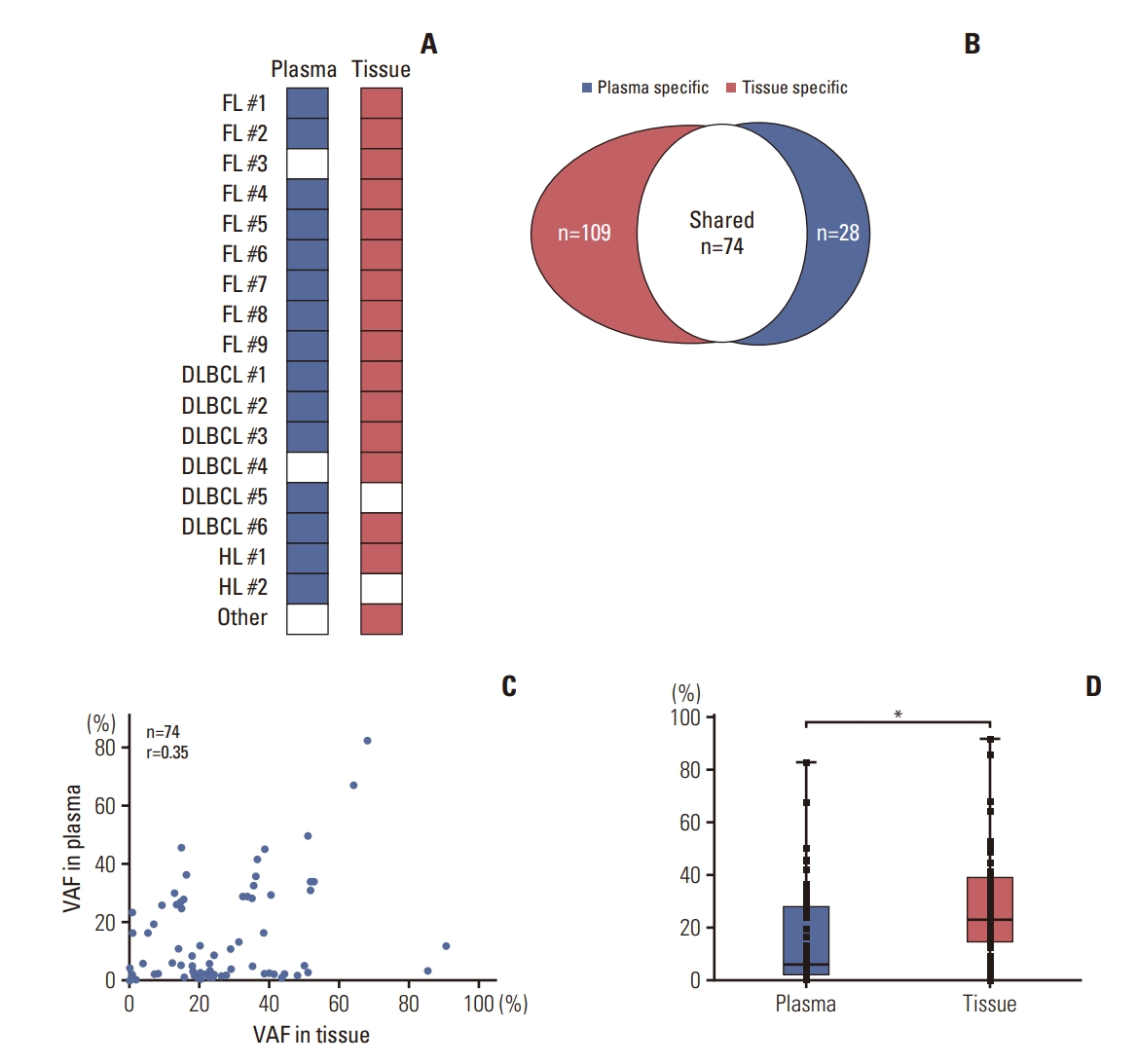
The average coverage depth was 1,555× (range, 847 to 2,299×) for tissue biopsy samples and 28,990× (range, 16,753 to 74,653×) for matched plasma samples. At least one mutation was detected in the majority of tissue biopsy samples (16/18, 88.9%) and plasma samples (15/18, 83.3%) across 112 target genes (Fig. 1A). The concordance between the genetic profiles of tissue and plasma, specifically regarding the presence or absence of genetic alterations in both sample types, was deemed acceptable for 13 out of 18 patients, corresponding to a rate of 72.2%. The medium number of variants per sample was 4 (range, 0 to 21) for plasma and 9 (range, 0 to 38) for tissue samples. A total of 183 somatic mutations were detected from the tissue samples, and 40.4% (74/183) of somatic mutations were observed in the matched 15 plasma samples (Fig. 1B). The variant allele frequencies (VAF) of each somatic mutation shared by the matched samples had a moderate degree of correlation (r=0.348) (Fig. 1C). The 74 somatic mutations shared by the matched samples had higher median VAF in the bone marrow (22.8%) than they did in the plasma (6.0%, p < 0.0001) (Fig. 1D). The somatic mutations detected only in tissue samples (3.8%; Mann-Whitney test p < 0.0001) or plasma (2.7%; Mann-Whitney test p=0.004) had lower median VAF than did the 74 shared somatic mutations in each sample type.
KMT2D was the most frequently mutated gene in matched tissue (56%, 10/18) and plasma (33%, 6/18) samples. The following genes were frequently mutated in tissue samples: CREEBP (39%, 7/18), BCL2 (28%, 5/18), KMT2C (22%, 4/18), and SPEN (22%, 4/18). The following genes were frequently mutated in plasma samples: IRF8 (28%, 5/18), BCL2 (22%, 4/18), IKZF1 (17%, 3/18), HIST1H1E (17%, 3/18), SOCS1 (17%, 3/18), SPEN (17%, 3/18), and CREEBP (17%, 3/18), followed by KMT2D.
3. Detection of ctDNA mutations in baseline samples
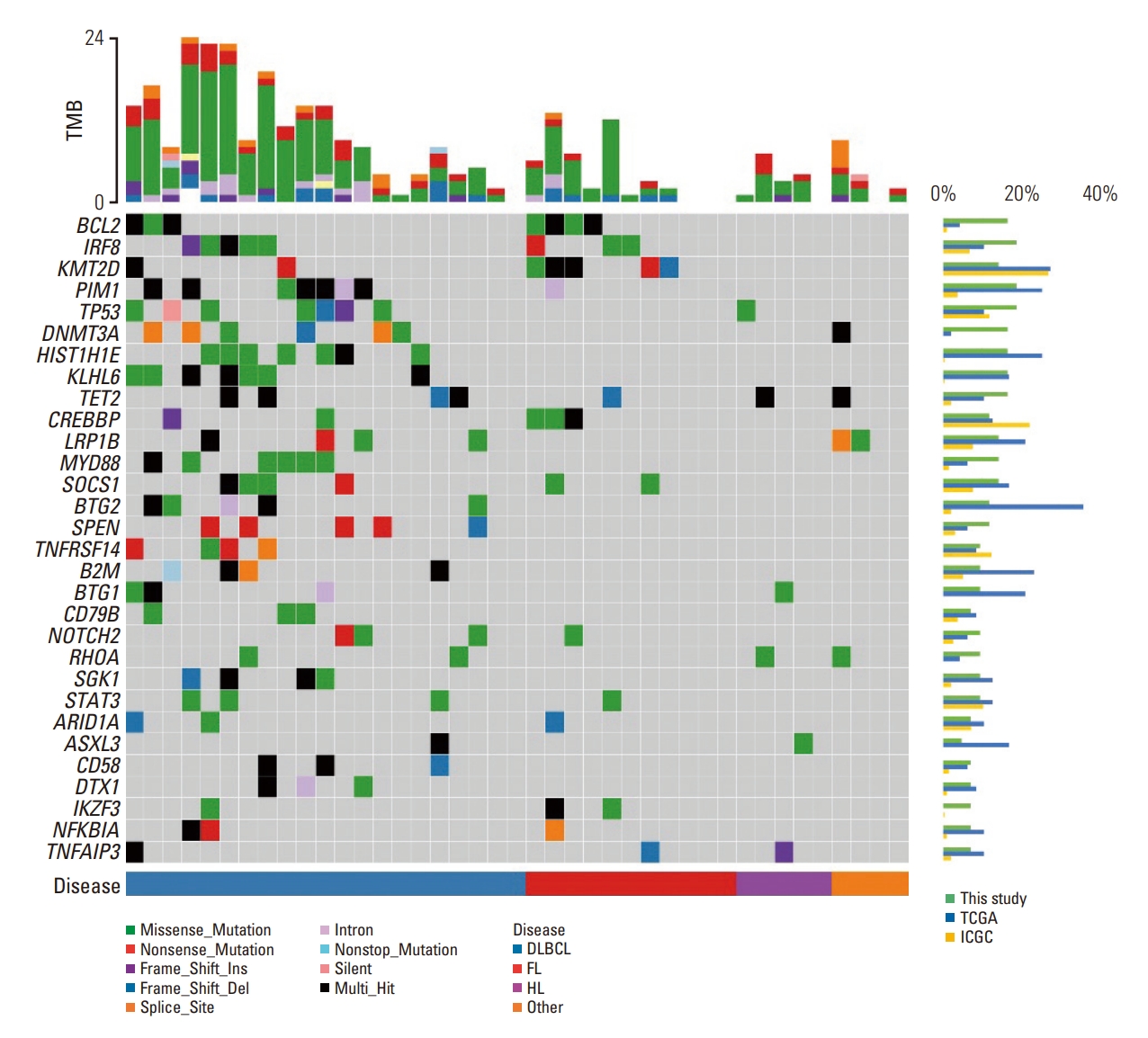
Forty-two baseline plasma samples from patients were sequenced. Three hundred four somatic mutations in 80 genes from 36 patients were identified (S2 Table). The frequently mutated genes included PIM1, TET2, BCL2, KMT2D, KLHL6, HIST1H1E, and IRF8 (Fig. 2). The median VAF of the detected mutations in the samples was 5.8%, and ranged 0.1% to 84.4%. The median number of variants per sample was 5.5 (range, 0 to 24). The overall detection rate was 85.7% (36/42). The detection rate of somatic mutations was highest in DLBCL (19/21, 90.5%). We conduct a characteristic analysis of tissue-paired samples from 18 individuals. (Table 2). The median age of patients was higher in patients with ctDNA mutation (52.0 years) than in patients without mutations (39.0 years). In addition, the median ctDNA concentration was higher in higher in patients with ctDNA mutation (6,180.0 pg/mL) than in patients without mutations (2,977.8 pg/mL). The ROC curve analysis for ctDNA concentration was able to distinguish between patients with ctDNA mutations and patients without ctDNA mutations with an estimated area under the curve of 0.819 (95% CI, 0.57 to 1.07). This analysis had a sensitivity of 88.9% and a specificity of 82.1% at the chosen cutoff point of > 4,506 pg/mL (S3 Fig.).
Survival analysis was performed on FL and DLBCL patients (S4 Fig.). The association between ctDNA mutation detection in the blood at diagnosis and PFS was analyzed in 33 patients, and the association with survival was analyzed in 31 patients whose survival status could be determined. The median follow-up period for all patients was 22.8 months, ranged 0.9-35.9 months. The median PFS duration was 24.8 months and 28.9 months for patients with ctDNA mutations and patients without ctDNA mutations, respectively (p=0.379) (S4A Fig.). The median OS duration was 24.8 months and 28.4 months for patients with ctDNA mutations and patients without ctDNA mutations, respectively (p=0.600) (S4B Fig.).
4. Comparison of plasma ctDNA concentrations
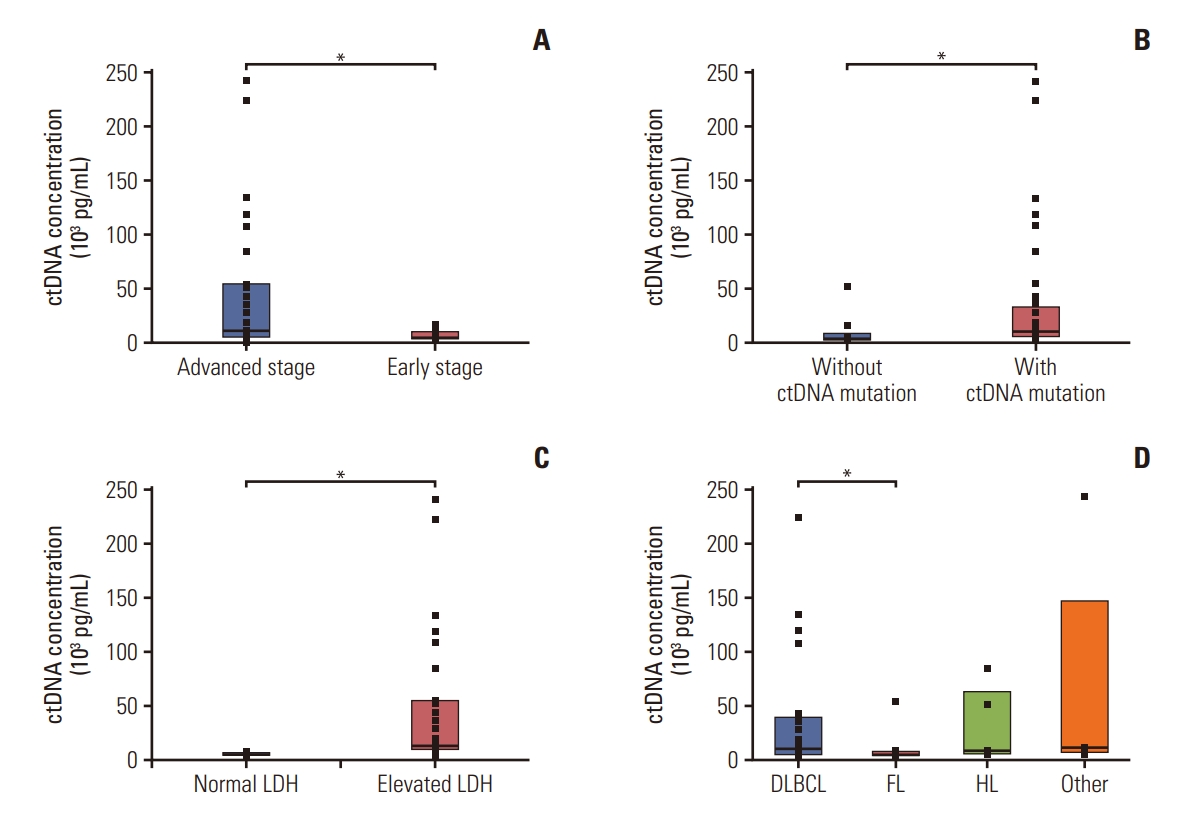
The correlation of ctDNA concentration and patients’ clinical status is depicted in Fig. 3A. The median plasma ctDNA concentration in patients was 9,321 pg/mL (n=42; range, 1,390 to 241,500 pg/mL). The median ctDNA concentration was greater in patients with advanced-stage disease (stage III, IV; 10,920 pg/mL) than it was in those with early-stage disease (stage I, II; 5,368 pg/mL) (p=0.014) (Fig. 3A). The median ctDNA concentrations of patients with bone marrow involvement (49,021 pg/mL) were greater than those without bone marrow involvement (8,848 pg/mL), although the difference was not statistically significant (p=0.073). The plasma samples with ctDNA mutations had higher median ctDNA concentrations (10,145 pg/mL) than did samples without mutations (3,965 pg/mL) (p=0.013) (Fig. 3B). In addition, the plasma samples with elevated lactate dehydrogenase (LDH) also had higher median ctDNA concentrations (12,180 pg/mL) than did those with normal LDH levels (4,328 pg/mL) (p < 0.001) (Fig. 3C). LDH was positively correlated with ctDNA concentration (r=0.41, p=0.007). Patients with B symptoms had higher ctDNA concentration (34,362 pg/mL) than did patients without B symptoms (12,407 pg/mL), although the difference was not statistically significant (p=0.263).
When we analyzed the correlation between ctDNA concentrations and lymphoma subtype, we found that the median ctDNA concentration of patients with DLBCL (10,605 pg/mL) was higher than among those with FL (5,069 pg/mL, p=0.010). Other than DLBCL and FL, the ctDNA concentration showed no relation to other lymphoma subtypes. The median ctDNA concentration of HL was 9,030 pg/mL, and that of other lymphoma subtypes was 11,673 pg/mL (Fig. 3D).
The detection rate of ctDNA mutations was high in DLBCL, but there was no statistical difference between the median VAF between the lymphoma subtypes. The median VAF was 5.8% in DLBCL, 5.1% in FL, 6.7% in HL, and 7.3% in other types of lymphomas (p=0.6344).
Discussion
There are limited studies that evaluate ctDNA in Korean patients with lymphoma. We designed an NGS panel and evaluated the clinical utility of our targeted sequencing platform for detecting plasma ctDNA mutations in lymphoma patients. This technique can be used for diagnosing and monitoring multiple lymphoma subtypes. We also validated this panel for unselected patients from a single institution, and confirmed its clinical utility.
The mutation detection rate of ctDNA was comparable to that of the tissue samples; 40.4% (74/183) of somatic mutations detected from tissue samples were also detected in plasma samples. When comparing it to the concordance rate observed in previous studies on lymphoma ctDNA, which reported the identification of 70% to 90% of biopsyconfirmed tumor mutations [14,15], the detection rate in this particular study may appear to be comparatively lower. However, within the tissue samples analyzed in this study, it was found that 55.2% (101/183) of the identified variants had an allele frequency of less than 20.0%, and 41.5% (76/183) had an allele frequency of less than 10%. These findings indicate a substantial presence of low-frequency variants. Moreover, it has been reported that variants with a VAF of less than 20% are challenging to detect in blood samples [7,11]. In this study, the concordance rate of variants with a VAF of 20% or higher was 53.7% (44 out of 82), which, although lower than previous reports, falls within an acceptable range according to our evaluation. Therefore, our test method can predict the status of genetic mutations in lymphoma tissues using blood samples. Furthermore, more genetic mutations and actionable gene mutations were found in ctDNA than were in matched tissue biopsy samples. This finding is in agreement with findings from other studies on solid cancers [16,17].
Within the 18 individuals with tissue-paired blood samples, patients were categorized as follows according to the somatic mutation detection: (1) 13 patients with tissue positive and plasma positive (tissue+/plasma+), (2) three patients with tissue positive but plasma negative (tissue+/plasma–), and (3) two patients with tissue negative but plasma positive (tissue-/plasma+). In all patients, mutations were detected in either tissue or blood samples.
In five patients with discordant results between tissue and plasma samples, three patients had ctDNA mutations in the plasma that were not detected in the tissue. Twenty somatic mutations were detected in a DLBCL #4 tissue sample with a VAF ranging 1.7% to 58.7%. Three somatic mutations were detected in an ENKTL #1 tissue sample. However, no somatic mutation was detected in the plasma samples. The tissue sample was taken from tonsil and nasal mucosa. Imaging showed that there was local disease involvement limited to the biopsy region with no evidence of distant metastasis. This result again suggests that it is difficult to detect mutations in cases of locally invaded lymphoma. In addition, only lymph node tissue from FL#3 had detectable mutations. FL is considered an indolent lymphoma. Plasma samples from patients with FL are expected to have around one-tenth of the ctDNA that is found in patients with DLBCL [18]. We similarly found that the mutation detection rate of ctDNA was lower in FL (75.0%, 9/12) than DLBCL (95.2%, 20/21). Mutations found in the FL#3 tissue samples have VAF of up to 27.2%. Mutations in tumor tissue samples with VAF less than 20% tend to go undetected in the plasma, which may explain the discrepant results between the tissue samples and plasma.
Some previous studies have suggested that the discordant mutations found in ctDNA are false-negative results from tissue biopsy samples that may be related to temporal and spatial heterogeneity [19,20]. This hypothesis suggests that the discordant mutations that are only found in tissue reflect low disease burden, which is why they are undetected in plasma. Realistically, not all lymphomas that invade multiple tissues can be tested at all tissue locations. Therefore, ctDNA analysis may be able to capture variants that are missed by single-site tumor biopsies.
Pretreatment ctDNA concentrations correlated with disease stage and LDH, which suggests that the levels are a putative biomarker of tumor burden in lymphoma [21]. In addition, the pretreatment ctDNA concentrations correlated with ctDNA mutation detection, and ROC analysis found that patients with higher tumor burden tended to have detectable plasma gene mutations.
In this study, at least one mutation was detected in 85.7% of pretreatment plasma samples from the lymphoma patients. This finding is consistent with prior studies that show that the mutation frequency varies from 52% to 99% in lymphomas [9-11,21-24]. This result also demonstrates the potentially universal applicability of ctDNA mutation detection. Moreover, the detection rate of ctDNA was highest in the DLBCL samples, and the VAF was also higher in DLBCL than it was in HL and FL. These findings are consistent with previous findings that the ctDNA fraction is higher in patients with aggressive lymphomas (such as DLBCL) than it is in patients with indolent lymphomas [1,15].
Mutation frequencies of most frequent 30 genes from ctDNA analysis are compared with the two sources of database, The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) in S5 Table. Data from projects on lymphoma patients, TCGA-DLBC from TCGA data-base, MALY-DE, NKTL-SG, and DLBC-US from the ICGC database are used. The mutation detection techniques used were different for each data, resulting in varying mutation frequencies. However, most of the mutation rate observed in this study fell within the range of the results from the two databases. In the case of MYD88 gene mutations, the frequency was relatively high in the results of this study. All six patients with detected mutations were diagnosed with DLBCL, and the seven identified mutations all showed a VAF of over 1.5%. Furthermore, six out of the seven mutations are commonly reported in DLBCL, including the MYD88 L265P mutation, which has a prevalence of around 16.5% [25] in DLBCL patients. Considering this, the mutation frequency of MYD88 in this study is reasonably acceptable.
There were frequent in the PIM1, BCL2, and KMT2D genes in the plasma samples from patients with DLBCL and FL in this study. The PIM1 mutation is not only thought to be involved in the pathogenesis of DLBCL [26], but also related to ibrutinib resistance in DLBCL [27]. We identified 17 nonsynonymous mutations in seven DLBCL patients and one FL patient with VAF ranging 1.9% to 20.3%. Mutations at the kinase domain of PIM1 are relevant to disease progression and treatment with PIM kinase inhibitors [28,29]. Frameshift mutations and missense mutations at the kinase domain are hypothesized to be oncogenic [29]. Most (15/16) of the missense mutations in this study located in the kinase domain and one (L2V) outside the kinase domain are thought to induce ibrutinib resistance. These results are consistent with previous studies. Frequent BCL2 mutations are observed in FL and DLBCL [30]. Mutations in BCL2 at diagnosis have been shown to confer poor prognosis in FL [31] and DLBCL [32]. Therefore, detecting BCL2 mutations is important. The KMT2D mutation is found frequently in B lymphoma; therefore, a novel target drug, such as a KDM5 inhibitor, has been suggested [33]. Five of the eight BCL2 mutations (62.5%) that were found in tissue samples were also detected in the plasma. Seven of the 13 KMT2D mutations found in tissue samples were also detected in the plasma. Additionally, one BCL2 and one KMT2D mutation were only found in the plasma. This finding suggests that the plasma can be an alternative to tissue sampling for risk stratification and drug selection when a tissue biopsy is not feasible.
There were some limitations in our study. The sample size is small to establish a conclusion. In addition, the follow-up period was too short to draw a conclusion regarding disease prognosis based on the ctDNA analysis. In our study, detection of ctDNA mutation did not show a statistically significant difference in OS and PFS, but further studies with larger sample size with longer follow-up period can induce more precise conclusion on the impact of mutation detection on patients’ prognosis. This study analyzed samples at diagnosis, but did not evaluate samples during treatment. Therefore, we are not able to evaluate the utility of our analysis in disease surveillance. The topic should be further explored through a larger, prospective design. By analyzing follow-up plasma samples, we expect to detect new mutations that are not identified at diagnosis in lymphoma patients with histologic transformation and relapse.
In conclusion, we designed a ctDNA panel for lymphoma patients that can be readily applied in clinical practice. Our design panel can detect many actionable gene mutations, including those at low frequency. Therefore, liquid biopsy can be applied clinically in the evaluation of lymphoma patients, especially in aggressive lymphoma patients.