Introduction
Primary central nervous system lymphoma (PCNSL) is a rare subtype of non-Hodgkin lymphoma (NHL), accounting for less than 2% of all NHLs, with most cases of PCNSL having diffuse large B-cell lymphoma (DLBCL) histopathology [1]. Because of the necessity to cross the blood-brain barrier (BBB), the induction treatment regimens for PCNSL differ from those for systemic DLBCL. Thus, chemotherapy containing high-dose methotrexate (HD-MTX) is the mainstay of PCNSL treatment [2]. However, relapse remains a significant issue, and one-third of PCNSL patients become refractory to salvage treatment [3]. Although previous mutation analyses have shown mutation profiles associated with the nuclear factor-κB (NF-κB) pathway, B-cell receptor activity, and immune evasion [4,5], information about PCNSL tumor biology and therapeutic targets is limited because brain biopsy is not always possible at diagnosis or relapse due to the risk of neurologic sequelae related to this invasive procedure.
Blood-based tumor diagnosis and surveillance through noninvasive blood sampling, or so-called liquid biopsy, is a rapidly advancing field in cancer research. Blood contains circulating tumor DNA (ctDNA), which is thought to be derived from tumor tissue and/or lysed circulating tumor cells [6]. The application of next-generation sequencing (NGS) for mutation analysis has allowed ctDNA-based tumor genotyping, and high-throughput sequencing of ctDNA could serve as a surrogate for sequencing the entire tumor genome [7]. Accordingly, ctDNA-based tumor genotyping might be a useful approach to providing genetic information about the primary tumor tissue that could identify potential therapeutic targets and allow rapid detection of relapse or residual disease in patients with PCNSL. Indeed, the possibility of applying liquid biopsy to brain tumor patients was suggested based on the observation that methylated tumor-specific DNA was present in the blood of glioma patients [8,9].
However, there are limited data about the relevance of ctDNA in the blood of PCNSL patients because it is not clear whether fragments of DNA from primary tumor cells can be shed into the bloodstream through the BBB. Actually, previous studies have suggested that plasma might not be optimal for ctDNA-based genotyping in primary brain tumors, including glioma, because of the BBB [10,11]. In a case report analyzing six patients with PCNSL, the plasma ctDNA analysis also had limited value compared with the analysis of cerebrospinal fluid (CSF)–derived ctDNA [12]. Thus, the feasibility of droplet digital polymerase chain reaction (PCR) with CSF-derived DNA for ctDNA analysis was reported in patients with PCNSL, rather than targeted sequencing of plasma cell-free DNA (cfDNA) [13,14]. Recently, the comparison of the CSF and plasma from 19 patients with central nervous system (CNS) and/or systemic lymphomas also showed the CSF ctDNA could better detect CNS lesions than plasma ctDNA using variant-specific droplet digital PCR for each mutation [15]. Likewise, the analysis of ctDNA mutation from CSF demonstrated the association with high-risk CNS involvement in patients with systemic DLBCL [16]. However, the CSF tapping through lumbar puncture is an invasive procedure that is not always easy to be done in outpatient clinics, especially for elderly and frail patients. Thus, there have been needs for blood-based ctDNA analysis because it might be more feasible and easily applicable than CSF analysis. Therefore, we performed targeted deep sequencing of plasma cfDNA in patients with PCNSL to explore the feasibility of plasma ctDNA mutation analysis in these patients and its association with their treatment outcomes.
Materials and Methods
1. Patients
We have conducted a single-center, prospective cohort study of adult patients with lymphoma since January 2017 (the Prospective Cohort Study for Lymphoma: ClinicalTrials.gov Identifier: NCT03117036). Patients admitted to our hospital for newly diagnosed or relapsed/refractory Hodgkin and NHL were eligible for this prospective cohort. Once patients were enrolled after providing written informed consent, they were treated after laboratory evaluation and monitored in a real-life context. At the same time, we collected and stored biological samples, including germline DNA, serum, and plasma cfDNA, for future research at our institute. As part of this prospective cohort study, we analyzed 42 patients with newly diagnosed PCNSL (n=39) or relapsed PCNSL (n=3) out of 751 patients enrolled between January 2017 and December 2018 (Fig. 1A). Plasma samples were collected at enrollment of the prospective cohort, after two cycles of induction chemotherapy, and at the end of treatment or at the time of progression (Fig. 1B). The tumor pathology was confirmed by two lymphoma pathologists (K.Y.H. and C.H), and all patients were evaluated for staging workup and risk stratification according to the guideline of the International Extranodal Lymphoma Study Group (IELSG), including physical examinations, complete blood cell counts, serum biochemistry, brain magnetic resonance imaging (MRI), bone marrow aspiration, ophthalmologic examination, and CSF cytology analysis. Treatment response was assessed as described by the International PCNSL Collaborative Group (IPCG) [17]. Thus, brain MRI was repeated during and after treatment to evaluate the response. For patients who achieved complete response, the follow-up evaluation with brain MRI was performed every three to four months as surveillance for relapse.
2. Sample preparation and cfDNA extraction
Whole blood samples were collected in Cell-Free DNA BCT tubes (Streck Inc., Omaha, NE). Plasma was prepared using three centrifugation steps at room temperature with increasing centrifugal force: 840 ×g for 10 minutes, 1,040 ×g for 10 minutes, and then 5,000 ×g for 10 minutes. After separation of plasma in the initial centrifugation, mononuclear cells were separated by Ficoll gradient centrifugation, and the granulocytes were purified from the pellet using Red Blood Cell lysis buffer (Qiagen, Santa Clarita, CA). Genomic DNA was isolated from the mononuclear cells using a QIAamp DNA mini kit (Qiagen). Plasma DNA was obtained from 2 to 5 mL of plasma using a QIAamp Circulating Nucleic Acid Kit (Qiagen). The AllPrep DNA/RNA Mini Kit (Qiagen) was used to purify genomic DNA (gDNA) from tissue samples. DNA concentration and purity were measured using a Qubit 2.0 Fluorometer (Life Technologies, Grand Island, NY). The fragment size distribution was measured using a 2200 TapeStation Instrument (Agilent Technologies, Santa Clara, CA).
3. Library preparation, sequencing, and data processing
Purified gDNA was sonicated (7 minutes, 0.5% power, intensity of 0.1, and 50 cycles/burst) into 150–200 bp fragments using a Covaris S2 (Covaris Inc., Woburn, MA). Tissue samples previously acquired for diagnosis were used to create reference libraries and were subjected to targeted sequencing. The tumor biopsy sample libraries were constructed using a SureSelect XT reagent kit, HSQ (Agilent Technologies), according to the manufacturer’s instructions. The mononuclear cell gDNA and plasma cfDNA libraries were created using a KAPA Hyper Prep Kit (Kapa Biosystems, Woburn, MA). Briefly, after end repair and A-tailing according to the manufacturer’s protocol, we performed adapter ligation at 4°C overnight using a customized adapter with molecular barcode (Integrated Device Technology, San Jose, CA). For the construction of libraries from biopsy specimens, hybrid selection was performed using customized probes targeting 426 lymphoma-related genes. To generate ultra-deep sequencing data at an affordable cost, we selected 54 genes according to the frequency of mutations in lymphoma. Thus, when we evaluated the coverage of these 54 genes for detecting somatic mutations across B-cell lymphomas using the COSMIC lymphoma database, 81.4% (848/972) of DLBCL patients were found to carry at least one mutation within the target regions of our panel. For sequencing of cfDNA in this study, capture probes for 54 genes were customized (Fig. 1B).
From the 54 genes, we designed baits targeting 617 regions covering 117,381 bases of the human genome. Total reads generated from cfDNAs and gDNAs were 39.6 and 33.5 million reads on average. After removing duplicates, we achieved 5,914.4× and 5,900.5× mean depth for cfDNAs and gDNAs, respectively. All data were aligned to the hg19 reference using BWAmem (v. 0.7.5, Wellcome Trust Sanger Institute, Cambridge, UK). Sequence Alignment/Map Format (SAM) files were converted to Binary SAM format using Samtools (v. 1.6, Wellcome Trust Sanger Institute). In preprocessing steps, Genome Analysis Tool Kit (GATK) (v. 4.0.0, Broad Institute, Cambridge, UK) and Picard (v. 2.9.4, Broad Institute) were used for quality controls of NGS data. The base recalibration process was performed by BaseRecalibrator and ApplyBQSR function in GATK software. Picard was used to identifying UID family in each group of PCR duplicates. After identifying UID family, we applied a home-built Python (v. 2.7.9, Python Software Foundation, Wilmington, DE) scripts to process the duplicate reads. We modified integrated digital error suppression methods by Newman et al. [18] and made scripts to perform the method, which combined in silico elimination of stereotypical background errors with a molecular barcoding strategy for the sensitive detection of ctDNA [19,20]. The parallel sequencing of matched white blood cells was performed to exclude mutations associated with clonal hematopoiesis and enable appropriate variant calling. During the processing, discordant pairs and off-target reads were filtered out. The variants that passed through the germline filter were designated as somatic mutations, and the filtering steps and sequencing metrics were summarized in Supplementary Methods and S1 Table [21]. The quantitative levels of ctDNA were measured as genome equivalents (GE) that were determined as the product of total cfDNA concentration and the maximal variant allele fraction (VAF) of somatic mutations.
4. Statistical analysis
The correlations between tumor burden and DNA concentration were calculated using the Pearson correlation coefficient, and the difference of cfDNA concentrations was compared by the Wilcoxon rank-sum test. Descriptive statistics were determined as proportions and medians, and the intergroup comparisons for categorical variables were assessed by Fisher’s exact test. Progression-free survival (PFS) was calculated as the date from diagnosis to disease progression or death related to any cause. Time to progression (TTP) was also calculated as the date from diagnosis to disease progression. Overall survival (OS) was defined as the period from the date of diagnosis to death or the last date of follow-up. Survival curves were described using Kaplan-Meier estimates and were compared between groups using the log-rank test. Statistical analyses were performed using an IBM PASW ver. 24.0 software program (IBM Corp., Armonk, NY).
Results
1. Patient characteristics
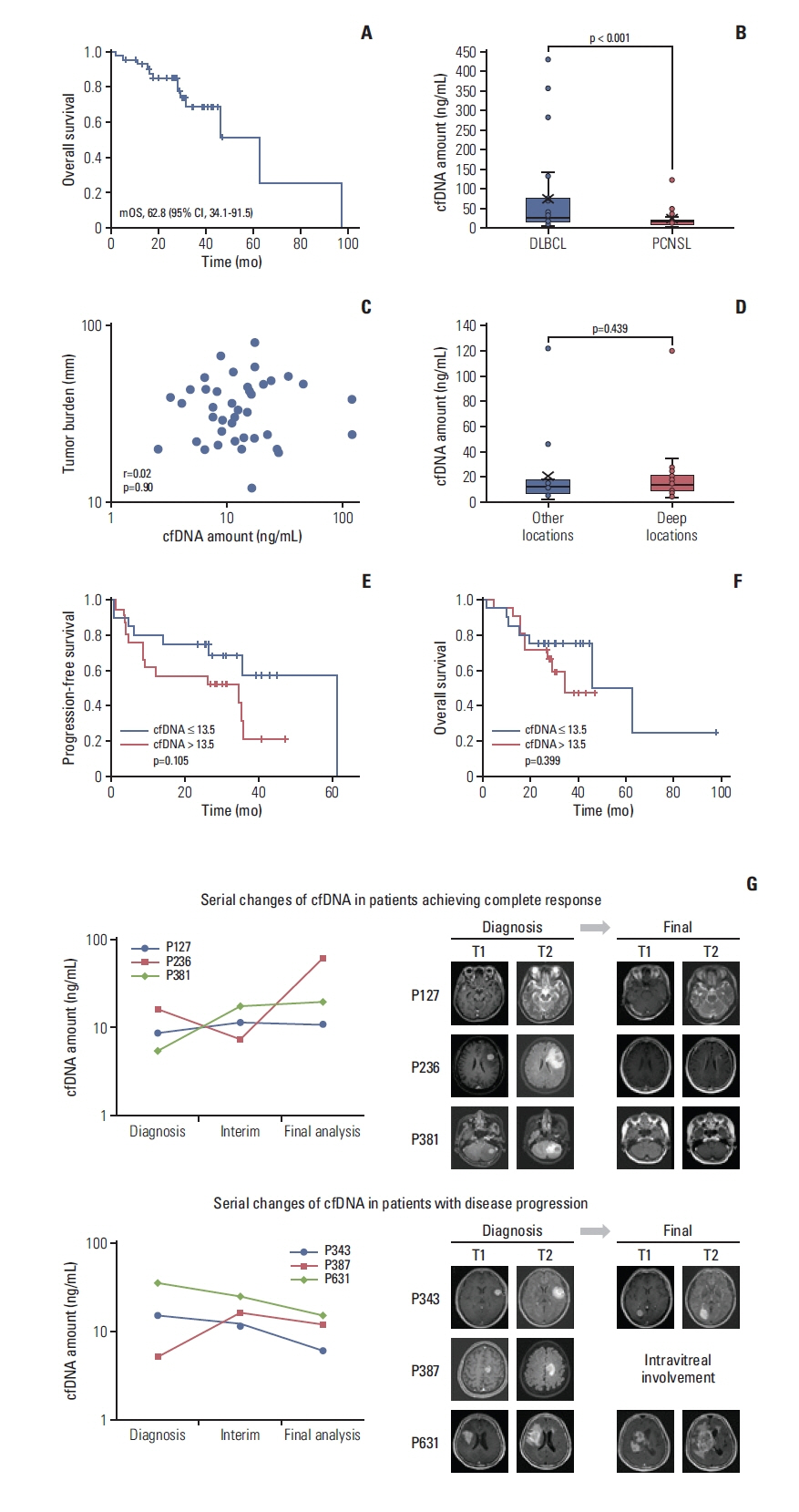
Of the 42 patients with PCNSL, 40 were diagnosed with PCNSL by stereotactic biopsy (n=5), navigation biopsy (n=25), or craniectomy (n=10), and the pathology of all tumors was DLBCL. Although the remaining two patients were diagnosed by brain MRI due to the deep location of the lesion within the brain parenchyma, their complete response to HD-MTX containing chemotherapy and their CSF findings suggestive of lymphoma cells supported the diagnosis of PCNSL. The patients’ median age was 52 years (range, 19 to 85 years), and the male (n=22) to female (n=20) ratio was approximately 1:1. The CSF analysis via lumbar puncture identified the presence of tumor cells in seven patients, and ophthalmologic examination identified one patient with intravitreal involvement. Based on the unfavorable features of the IELSG (age > 60 years, Eastern Oncology Cooperative Group performance ≥ 2, elevated serum lactate dehydrogenase, elevated serum protein level, and involvement of deep brain structures including periventricular regions, basal ganglia, corpus callosum, brain stem, and cerebellum), more than half the patients (n=25) had 2–3 unfavorable features and were classified as within the intermediate-risk group (Table 1). All patients with newly diagnosed PCNSL (n=39) were uniformly treated with rituximab, HD-MTX, vincristine, and procarbazine as an induction treatment after diagnosis, and the remaining three patients who relapsed after HD-MTX–containing chemotherapy received ifosfamide, carboplatin, and dexamethasone as salvage chemotherapy. As a consolidation treatment, autologous stem cell transplantation (n=8), high-dose cytarabine chemotherapy (n=18), or whole-brain radiotherapy (n=10) were performed (Table 1). During the follow-up, eight patients died because of disease progression (Fig. 2A).
2. Analysis of plasma cfDNA
The median cfDNA concentration in the 42 patients was 13.5 ng/mL (range, 2.6 to 121.6 ng/mL), which was significantly lower than the previously reported level in patients with systemic DLBCL (median, 25.5 ng/mL; range, 4.8 to 427.6 ng/mL; p < 0.001) (Fig. 2B) [21]. The concentration of cfDNA was not significantly related to the tumor volume as estimated by the long axis of the tumor mass on brain MRI (r=0.02, p=0.90) (Fig. 2C). The involvement of deep brain structures such as periventricular regions, basal ganglia, brainstem, and cerebellum was not associated with the concentration of cfDNA (p=0.439) (Fig. 2D). Hence, when we dichotomized patients into low- and high-cfDNA based on the median concentration of cfDNA, the pretreatment concentration of cfDNA was not significantly associated with the TTP (S2A Fig.), PFS, and OS (Fig. 2E and F). Likewise, serial changes in cfDNA did not show any significant association with the treatment outcome or disease relapse (Fig. 2G).
3. Targeted sequencing and mutation profiles of ctDNA
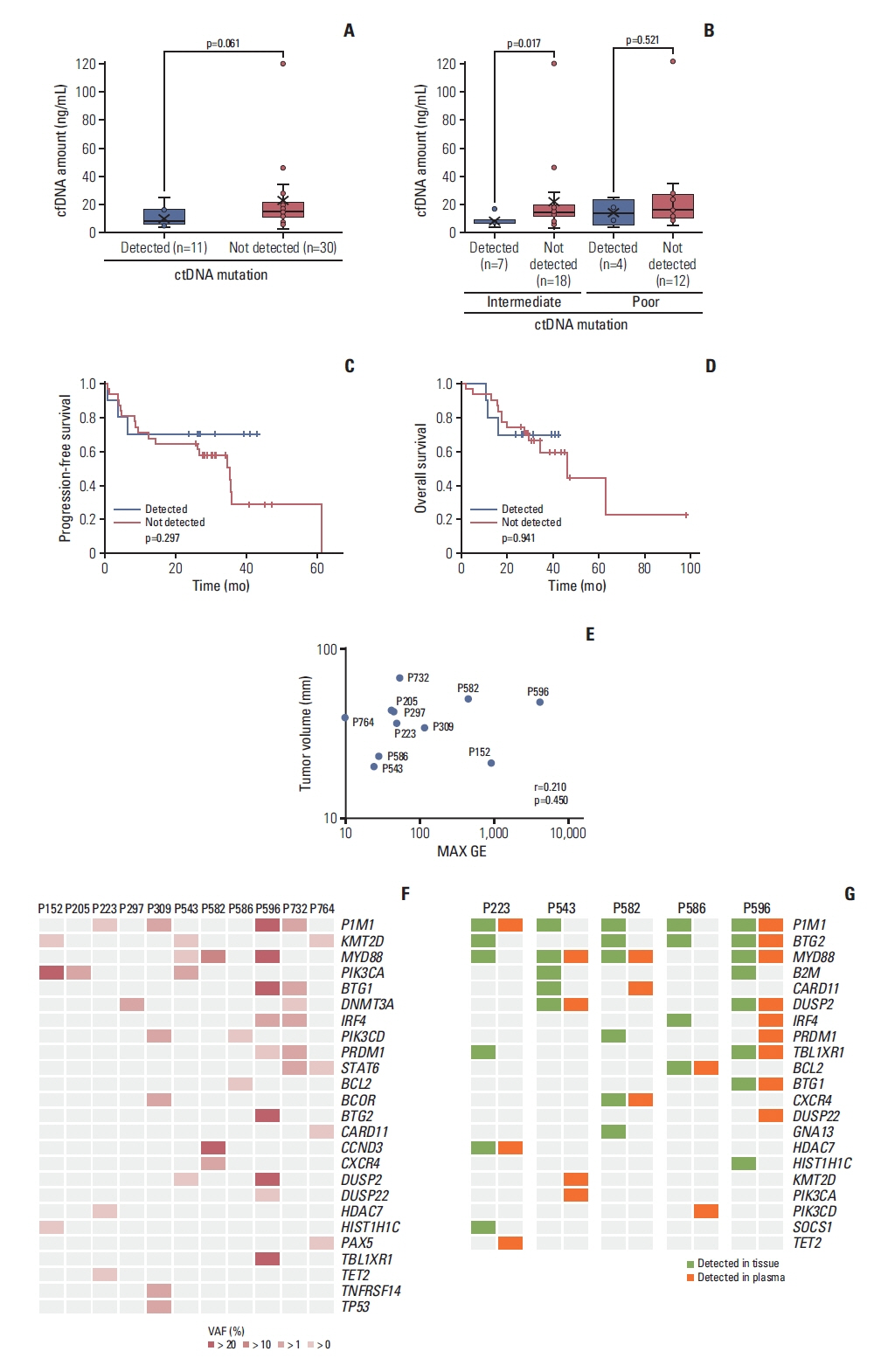
Targeted sequencing could be performed on the cfDNA samples of 41 of 42 enrolled patients, the exception being one patient with relapsed PCNSL whose isolated DNA was of poor quality. Somatic mutations within the target genes were detected in 11 patients (11/41, 27%) but not in the remaining 30. The median concentration of cfDNA in the 11 patients in whom a mutation was detected was not significantly different from that in the remaining 30 patients (p=0.061) (Fig. 3A). The detection of a mutation was not related to the concentration of cfDNA within the poor-risk groups of IELSG, whereas the concentration of cfDNA was higher in 18 patients who did not show mutations than patients whose mutation was detected (Fig. 3B). Out of 42 patients, dexamethasone was empirically administered in 33 patients prior to the induction chemotherapy. However, the use of dexamethasone did not influence the amount of cfDNA and detection of ctDNA mutation (data not shown). The comparison of survival outcomes between patients with and without detected mutations showed no significant difference in TTP (S2B Fig.), PFS, and OS (Fig. 3C and D). Similarly, there was no significant association of maximum GE with tumor volume in the 11 cases where ctDNA mutation was detected (r=0.210, p=0.450) (Fig. 3E). Mutation profiles of these 11 cases varied from one to multiple somatic mutations per patient (Fig. 3F). Of the genes in which mutations were detected, PIM1 mutation was the most frequently found (4/11, 36.4%), and the mutations of KMT2D, PIK3CA, and MYD88 were each observed in three patients (3/11, 27%). Mutations of BTG1, CARD11, DNMT3A, IRF4, PIK3CD, PRDM1, and STAT6 were each found in two patients, whereas mutations of BCOR, BTG2, CXCR4, DUSP2, DUSP22, HIST1H1C, PAX5, TBL1XR1, TET2, TNFRSF14, and TP53 were each detected in only one patient (Fig. 3F). The VAF of these mutations in plasma ctDNA varied between patients: the majority of patients showed low VAF for each gene, with three exceptions (Fig. 3F): P596 (PIM1, MYD88, BTG1, and BTG2), P152 (PIK3CA), and P582 (CCND3, MYD88). In addition, the VAFs of KMT2D, PRDM1, IRF4, and STAT6 were much lower than those of other genes. Next, we compared the mutation profiles identified in plasma ctDNA with those of tumor tissue. In the five patients with tumor tissue samples available for targeted sequencing, the mutation profiles of the primary tumor showed a higher number of somatic mutations than the mutation profiles of plasma ctDNA (n=29 vs. n=13) (Fig. 3G). Of these 29 somatic mutations, 13 were concordantly detected in plasma ctDNA, giving a detection sensitivity of 45%. The median VAF of plasma ctDNA mutations (4.3%; range, 0.5% to 51.2%) was also lower than that of tumor tissue (25.1%; range, 2.3% to 64.7%).
4. Association of plasma ctDNA mutation profiles with treatment outcome
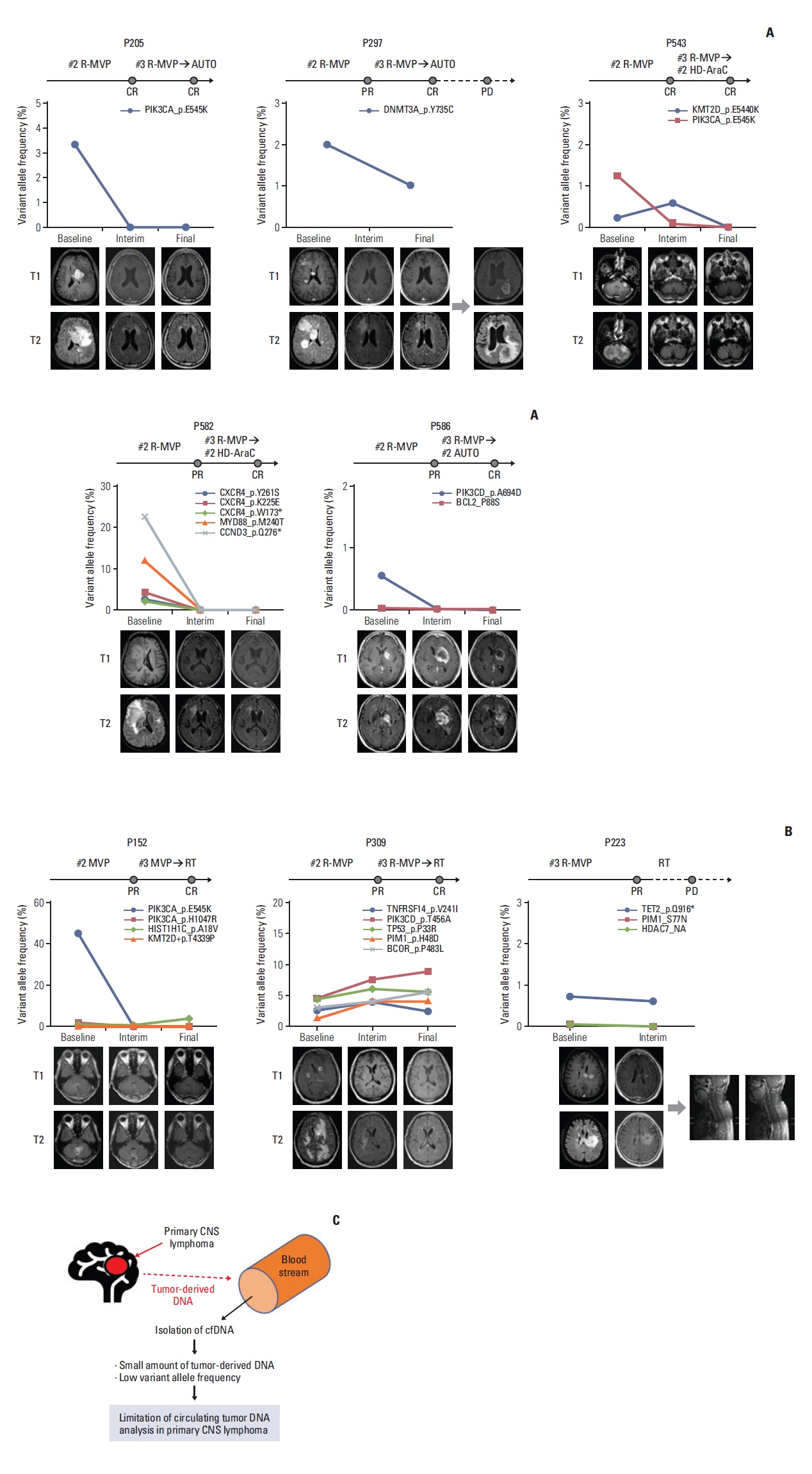
One patient harboring pretreatment plasma ctDNA multiple mutations, including MYD88, PIM1, BTG1, and BTG2, experienced early progression resulting in death (P596) (Fig. 4A). On the other hand, the other patient with multiple mutations involving KMT2D, STAT6, CARD11, and PAX5 was alive even after relapse (P764) (Fig. 4B). When survival outcomes of 11 patients whose ctDNA mutations were detected at diagnosis were compared, there was no significant association between high plasma ctDNA mutation burden and poor survival (Fig. 4C). However, patients with relatively low plasma ctDNA mutation burden seemed to be maintained complete response with no evidence of disease progression (P205, P543, P586) (Fig. 4C). When the serial tracking of plasma ctDNA mutation profiles through treatments were performed, four patients showing the complete disappearance of ctDNA mutations at the final response evaluation achieved complete response at the end of treatment and maintained their response without any evidence of relapse (P205, P543, P582, P586) (Fig. 5A). However, out of five patients who achieved complete response at the end of treatment, the one patient (P297) showed the persistence of detected DNMT3A mutation at the end of treatment. This patient eventually relapsed during follow-up, and died due to disease progression (Fig. 5A).
On the other hand, one patient showed a new mutation in the interim analysis but did not show any evidence of relapse at the time of analysis (P152) (Fig. 5B). Likewise, another patient receiving hemodialysis because of chronic renal failure showed the persistence of mutations at the interim and end of treatment analyses (P309) (Fig. 5B), although the patient maintained the response at the time of analysis. By contrast, the other patient harboring a persistent TET2 mutation at the interim and end of treatment analyses showed a late relapse during follow-up (P223) (Fig. 5B). In S3 Table, we presented serial sequencing results before and after chemotherapy for all patients for which ctDNA was detected.
Discussion
In this study, we investigated the clinical relevance of plasma ctDNA-based mutation analysis in 42 patients who were consecutively enrolled in our prospective cohort. Considering that the feasibility of blood-based genotyping is unclear in patients with a brain tumor because of the BBB, we first measured the concentration of cfDNA in the blood of patients with PCNSL and analyzed its role as a biomarker for predicting tumor burden and treatment outcome. We observed that the concentration of cfDNA in PCNSL patients was much lower than that in patients with systemic DLBCL (Fig. 2B). Furthermore, tumor volume and tumor location were not associated with the concentration of cfDNA (Fig. 2C and D). Hence, the pretreatment cfDNA concentration and the serial changes in concentration during treatment failed to predict the treatment outcomes (Fig. 2E–G). These findings implied a lack of clinical usefulness of cfDNA concentration as a liquid biopsy-driven biomarker in patients with PCNSL.
However, plasma ctDNA-based mutation analysis detected somatic mutations in genes including KMT2D, PIK3CA, PIM 1, BTG1, CARD11, DNMT3A, IRF4, MYD88, PIK3CD, PRDM1, and STAT6, and these mutations were consistent with those identified in a previous study reporting the detection of somatic mutations related to NF-κB activation (MYD88, PIM1, IRF4, CARD11, PRDM1), apoptosis (GNA13, TP53, CDKN2A, BCL2) and B-cell receptor signaling (CD79B, ITPKB) in ctDNA of PCNSL [22]. In our study, patients having a low pretreatment plasma ctDNA mutation burden maintained their remission status (P205, P543, P586) (Fig. 4C), and patients with the disappearance of somatic mutations that were initially found in the pretreatment sample were also alive without any evidence of relapse (P205, P543, P582, P586) (Fig. 5A). Furthermore, one patient with the perstence of detected ctDNA mutation eventually relapsed (P297) (Fig. 5A). These findings might suggest the potential role of ctDNA mutations in blood for monitoring disease status and predicting relapse of patients with PCNSL. However, the number of patients whose mutations were detected was too small to draw a robust conclusion. Actually, the other patients with persistently detected ctDNA mutations maintained their response regardless of ctDNA mutations (P152 and P309) (Fig. 5B). Among them, one patient who received hemodialysis because of chronic renal failure showed persistence of a somatic mutation in plasma ctDNA even after the achievement of complete response (P309) (Fig. 5B). Although this could not be fully explained, it could be associated with impaired clearance related to renal failure because a previous study suggested that the detection of ctDNA in urine could reflect the loss of ctDNA from the kidney [23]. Another case showed a persistently detectable TET2 mutation and ultimately relapsed during follow-up (P223) (Fig. 5B). Although TET2 mutations are mainly associated with T-cell lymphomas rather than DLBCL, genome-sequencing studies have shown that around 10% of DLBCL have somatic mutations of TET2 [24–26]. Furthermore, TET2 mutation was reported to promote B-cell lymphomagenesis [27]. However, the meaning of TET2 mutation should be more clarified in future studies with a larger number of patients because it is still not clear in the interpretation of a TET2 mutation of PCNSL.
In this study, the detection of mutations in plasma ctDNA was possible in only 11 of 41 patients (27%) whose cfDNA was available for targeted sequencing. This performance was inferior to that reported for systemic DLBCL in our previous study, even though we used the same platform for targeted sequencing [21]. This low rate of mutation detection was consistent with a previous study of various brain tumors, including medulloblastoma (20%), glioblastoma (27%), and CNS lymphoma (32%) [28]. Furthermore, when we compared the mutation profiles of plasma ctDNA with those of primary tumor tissue, the frequency of MYD88 mutations detected in plasma ctDNA (2/5, 40%) was lower than that in primary tissue (4/5, 80%). Although we could compare data for only five patients, the concordance rate between tissue and plasma ctDNA was less than 50%. These results imply that plasma ctDNA has limited value for providing information about the mutation profiles of primary tumors in PCNSL. In fact, previous studies also reported a relatively low frequency of MYD88 mutations (around 35%) in plasma ctDNA compared with that in primary tumor tissue of PCNSL [14,28]. This limited sensitivity of plasma ctDNA-based mutation analysis in PCNSL might be related with the intrinsic feature of CNS tumors, including the limited transfer of cfDNA from the CNS or CSF to the bloodstream. Actually, the concentration of tumor-derived DNA in PCNSL was much lower than that in systemic DLBCL, and it might be related with that the BBB interferes with the shedding of tumor-derived DNA into the bloodstream (Fig. 2B). Although the concentration of cfDNA did not differ significantly between patients with and without mutations detected in plasma ctDNA (Fig. 3A), it is possible that the concentration of tumor-derived DNA might be too small to allow targeted sequencing (Fig. 5C). Consistent with this, the VAF of mutations in plasma ctDNA of our study was extremely low in the majority of cases (Fig. 3F). Another confounding factor for interpreting the role of plasma ctDNA in patients with PCNSL could be the presence of cfDNA derived from other cells because plasma cfDNA may carry noncancerous mutations such as clonal hematopoiesis of indeterminate potential mutations, and such mutations could accumulate with age. Actually, our study showed the detection of some mutations in the plasma that were not identified in tumor tissue (Fig. 3G). Although we analyzed mononuclear cell gDNA in parallel to discriminate such noncancerous mutations from tumor somatic mutations in ctDNA, this may not have eliminated all noncancerous somatic mutations. Lastly, the limited number of genes could influence the low detection rate of tumor-derived ctDNA mutations in plasma. Although we evaluated the performance of our gene panel in terms of coverage for somatic mutations using the COSMIC databases, some patients might carry any mutations of genes that were not included in our panel.
Therefore, these factors still remain as hurdles to the clinical application of plasma ctDNA-based mutation analysis in patients with PCNSL (Fig. 5C). The comparison of ctDNA analyses using matched blood and CSF samples would have clarified the effect of limited transfer of cfDNA from the CNS or CSF to the bloodstream on the limited value of plasma ctDNA analysis in patients with PCNSL. However, we could not perform targeted sequencing of CSF-derived ctDNA because there were no archived CSF samples during the study period of the prospective cohort. This remained as the limitation of our study, and the efficacy of an analytical platform must also be improved to enhance the detection rate of somatic mutations from small amounts of genetic material with extremely low VAF. Since the allele frequency of ctDNA mutations is overlapped with the range of the NGS technology error rate, the use of molecular barcode technology might be a solution for the problem. However, a more sensitive barcode filtering technology should be required. Furthermore, the preparation of samples such as isolation of cfDNA should be optimized to improve the extraction yield and the sensitivity for detecting ctDNA mutation at a very low allele frequency.
In conclusion, we conducted the first study to investigate the feasibility of plasma ctDNA-based mutation analysis in prospectively enrolled patients with PCNSL. Although some patients achieving complete response showed the disappearance of plasma ctDNA mutations after treatment, the number of patients was too small to draw a conclusion regarding its usefulness. Considering the low sensitivity to detect mutations in our results, the clinical application of plasma ctDNA mutation analysis has limitations for surveillance and predicting treatment outcomes of PCNSL. The efficacy of analytical platforms should be improved to overcome current hurdles associated with the application of plasma ctDNA analysis for patients with PCNSL.