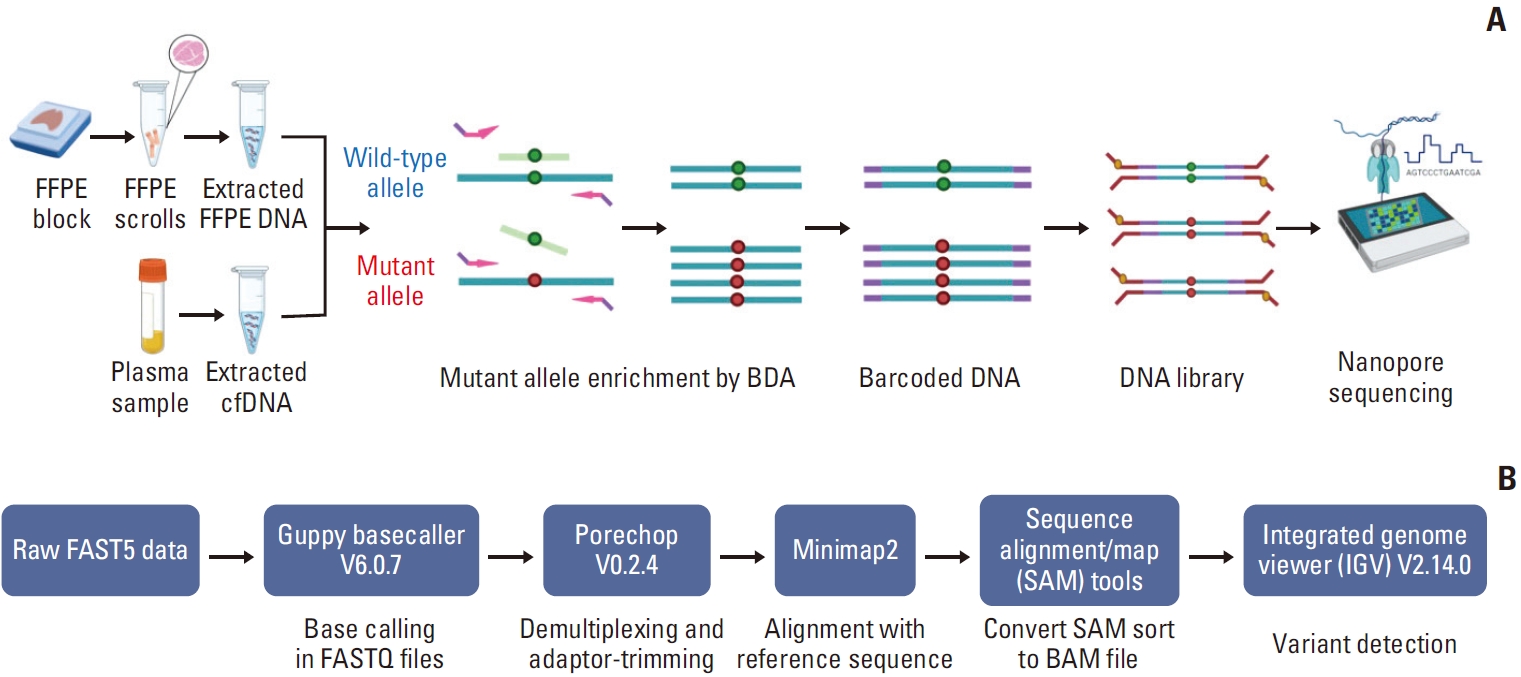
Introduction
The epidermal growth factor receptor (EGFR) mutation is a common oncogene driver accounting for 50% of patients with non–small cell lung cancer (NSCLC) in East Asia [1]. Nowadays, EGFR tyrosine kinase inhibitors (EGFR-TKIs), namely erlotinib, gefitinib, and osimertinib, are used as the standard targeted therapy in NSCLC patients with EGFR mutations [2]. The 19Del and L858R EGFR mutations respond effectively to erlotinib and gefitinib treatment, inducing favorable outcomes [3,4]. However, the T790M mutation confers resistance to gefitinib or erlotinib therapy but remains sensitive to osimertinib, the third-generation EGFR-TKI [3,5]. Therefore, the detection of EGFR mutations is a standard routine testing to guide personalized treatment in clinical practice [2-4].
Cobas real-time polymerase chain reaction (PCR) is a standard method for detecting EGFR mutations in formalinfixed, paraffin-embedded tissues (FFPE) with a sensitivity of at least 5% mutation levels, which is insufficient to detect cell-free DNA (cfDNA) containing low-level mutated DNA at less than 1% [6]. Instead, droplet digital PCR (ddPCR) is a technique that partitions the PCR reaction mixture into thousands of water-in-oil droplets, allowing for absolute quantification of target DNA with higher sensitivity than real-time PCR [7]. Therefore, ddPCR is used as the standard method for detecting EGFR mutations in cfDNA from blood [6,7]. These methods use an allele-specific forward primer designed to have a nucleotide at the 3’ end perfectly matched with the mutant target [8]. However, this approach still has crossreactivity with wild-type alleles [9]. To improve allele discrimination, the blocker methods, including peptide nucleic acid (PNA), locked nucleic acid (LNA), and blocker displacement amplification (BDA), are used to avoid wild-type allele amplification [8,10].
PNA is a synthetic peptide polymer analog with nucleic acid bases as side chains, which can form hydrogen bonds to complementary DNA targets [11]. However, it lacks 3′OH, preventing DNA polymerase extension [11]. LNA is bridged nucleic acid that locks ribose moiety in the 3′-endo conformation, which prevents the cleavage mismatch nucleotide and enhances mismatch discrimination [12]. BDA uses a DNA oligonucleotide probe with internal three-carbon spacer, which can bind to wild-type DNA and prevent the binding of PCR primer [13,14]. Hence, BDA can limit the amplification of the wild-type alleles and effectively enrich low variant allele frequency (VAF) [9,13].
To identify the amplified product, the second-generation sequencing such as Illumina is commonly used [14]. However, these instruments are expensive and only available in centralized laboratories [14]. Recently Nanopore sequencing, a third-generation long-read sequencing, has gained more attention [14]. Its sequencer is portable and more affordable, allowing its usage in low resource-setting areas [15,16]. To combine Nanopore sequencing with PCR enrichment approach, BDA rather than PNA and LNA was chosen because it uses oligonucleotide to block the amplification of wild-type allele [13]. In contrast, LNA binds tightly to mutant alleles, resulting in amplification of mutant alleles [12,13]. However, this amplicon contains LNA, which is not suitable for Nanopore sequencing [13]. For PNA, it is a peptide-based nucleotide analog, which is more difficult and expensive to synthesize than LNA and BDA probes [8,10].
Materials and Methods
1. Samples
The H1650 has 19Del mutation (E746_A750) in exon 19. The H1975 has T790M and L858R mutations in exons 20 and 21, respectively. Therefore, H1650 was used as a positive control for detecting EGFR 19Del, while H1975 was used for detecting EGFR T790M and L858R mutations. These cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco’s modified Eagle medium low glucose (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum and 1% antibioticantimycotic (Gibco, Gaithersburg, MD) at 37°C in a humidified incubator with 5% CO2.
The genomic DNA from H1650 and H1975 were extracted by GenUP gDNA Kit (Biotechrabbit, Berlin, Germany), according to the manufacturer’s instructions. The quantity of DNA was subsequently measured using the Nanophotometer C40 (Implen, Munchen, Germany).
A cohort of 65 FFPE DNA and 50 cfDNA samples from NSCLC patients with known EGFR mutations by Cobas real-time PCR and ddPCR, respectively, were obtained from the Chula GenePRO center, Faculty of Medicine, Chulalongkorn University under the approval of the Institutional Review Board of Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand, IRB No. 0299/65 (S1 Table for the patient’s age, sex, cancer types, and other specimen details).
2. Preparation of positive controls for detection
The EGFR gene in exons 19, 20, and 21 covering 19Del (position chr7:155728-155864), T790M (position chr7:162339-162483), and L858R (position chr7:172777-172914), respectively, were PCR-amplified using genomic DNA from the cancer cell lines. All amplicons were purified by QIAquick PCR Purification kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Then, all purified samples were ligated to pGEM-T easy vector (Promega, Madison, WI). Ligation reaction, a total volume of 10 µL contained 2.5 µL of 5× rapid ligation buffer, 1 µL of T4 DNA ligase, 1 µL of 50 ng pGEM T easy (TA) vector, and 5.5 µL of DNA insert. Subsequently, all ligated products were incubated at 22°C for 60 minutes and transformed into Escherichia coli DH5α. The recombinant plasmids containing wild-type and mutant EGFR were extracted by PureDirex Plasmid miniprep kit (BIO-HELIX Co., Ltd., Taipei, Taiwan), following the instructions provided by the manufacturer. The EGFR gene sequence in the recombinant plasmids was verified by Sanger sequencing, as shown in S2 Fig.
3. BDA assay
The three primer pairs and blocker probes were used to amplify EGFR exon 19 (position chr7:155728-155864), exon 20 (position chr7:162339-162483), and exon 21 (position chr7: 172777-172914) covering each mutant position (Table 1, S3 Fig.). Forward primers were placed close to the mutant nucleotides, with 6 to 14 nucleotides overlapping with blocker probe sequences. Reverse primers were designed to prevent dimer formation between the primers and blockers. Subsequently, the 5’ end of primers were tailed with adapter nucleotide for tagged with barcode for Nanopore sequencing. The blocker probes were designed to complement with wild-type allele. The 3’ end of the blocker was modified into a 3-carbon spacer to prevent polymerase extension.
In the BDA-based PCR reaction, a total volume of 10 µL contained 1 µL of DNA template, 1 µL of 1× Standard Taq Reaction Buffer (New England Biolabs, Hitchin, UK), 0.2 µL of 200 µM dNTPs mixed, 0.1 µL of 10 µM of each primer (U2Bio, Seoul, Korea), 1 µL of 10 µM blocker (U2Bio), 0.125 µL of 100 U Taq DNA polymerase, and 6.475 µL of DEPC treated water. The following PCR settings were used for amplification: initial denature at 95°C for 10 minutes, 35 cycles of amplification (95°C for 10 seconds, 58°C for 2 minutes, 68°C for 30 seconds) and final extension at 68°C for 10 mintues. Then, the amplified products were tagged with different barcodes.
For the PCR barcoding step, a total volume of 60 µL contained 3 µL of amplified products, 1× Standard Taq Reaction Buffer (New England Biolabs), 200 µM dNTPs mixed, 0.2 µM of each barcode primer from PCR Barcoding Expansion 1-96 (EXP-PBC096) kit (Oxford Nanopore Technologies, Oxford, UK), 0.5 U Taq DNA polymerase, and DEPC treated water. The PCR conditions included initial denaturation at 95°C for 10 minutes, 10 cycles of amplification (95°C for 10 seconds, 55°C for 1 minute, 68°C for 1 second), and final extension at 68°C for 5 minutes. After that, the amplified samples were purified by QIAquick PCR Purification kit (Qiagen), according to the manufacturer’s instructions. Finally, the concentrations of purified products were measured by Qubit HS DNA Assay Kit (Invitrogen, Carlsbad, CA) and Qubit 4 fluorometer (Thermo Fisher Scientific, East Grinstead, UK).
4. DNA library preparation and sequencing
All samples were pooled equally to final amount of 1 µg DNA in distilled water. The adapters were ligated to the pooled samples using SQK-LSK114 kit (ONT, London, UK). The DNA library was loaded into a flow cell ver. R10.4.1 (FLO-MIN114) and sequenced by MinION platform. Approximately 10,000 and 30,000 reads per sample were sequenced for FFPE and cfDNA, respectively.
5. Bioinformatics
The raw read data in the FAST5 files were converted to the FASTQ files by Guppy basecaller software ver. 6.0.7 (https://github.com/asadprodhan/GPU-accelerated-guppy-basecalling). To mitigate the Nanopore sequencing error background, we used super accuracy Phred quality score (Q score) ≥ 15 settings for base calling that includes an error rate of 3.16% or less. Thus, the sequencing used minimum read depth at 1,000 to ensure base accuracy. Then, the filtered reads were checked for quality control of sequencing data by MinIONQC (https://github.com/roblanf/minion_qc) before demultiplexed and adapter-trimmed by Porechop Ver. 0.2.4. After that, total demultiplexed reads were rechecked in quality and length of sequenced read by FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and then aligned with the human reference genome (GRCh38) using the Minimap2 aligner (https://github.com/lh3/minimap2) to generate SAM files. Then, the files were sorted and converted to BAM files by Samtools (https://www.htslib.org/). The Integrated Genome Viewer (IGV) program ver. 2.14.0 (https://software.broadinstitute.org/software/igv/) was utilized to visualize mutations and calculate VAF.
6. Identification of EGFR mutations status
The Clinical and Laboratory Standards Institute (CLSI) EP17-A guidelines were used to determine limits of blank (LOB) and limits of detection (LOD) [17,18]. In this study, LOB stands for the lowest VAF from sequencing error. The LOD stands for the lowest enriched VAF from sequencing.
Wild-type patients from FFPE (n=5) and cfDNA (n=9) samples were down-sampled to calculate sequencing error based on the %VAF. The enriched VAF studies were conducted by diluting each mutant EGFR ranging from 5%, 2.5%, 1%, 0.5%, and 0.1%, which mimic low VAF in clinical specimens. Samples with %VAF higher than the determined cutoff were classified as mutants. Conversely, samples with %VAF lower than the cutoff were classified as wild type.
Sequencing error=meanwild-type sample+1.645 (SDwild-type sample) (1)
Enriched VAF=sequencing error+1.645 (SDlow concentration sample) (2)
7. Assessment of the performance in clinical specimens
To assess the performance of this technique, the results from FFPE DNA and cfDNA samples were then compared with the real-time PCR and ddPCR, respectively. The analytical sensitivity was calculated by TP/(TP+FN). The specificity was calculated by TN/(TP+FP), and concordance rate was calculated by (TP+TN)/(TP+FP+FN+TN). TP, TN, FN, and FP indicate true-positive, true-negative, false-negative, and false-positive.
Results
1. Developing blocker displacement amplicon Nanopore sequencing to detect EGFR mutations
(1) Evaluation of the assay for detecting cancer cell lines
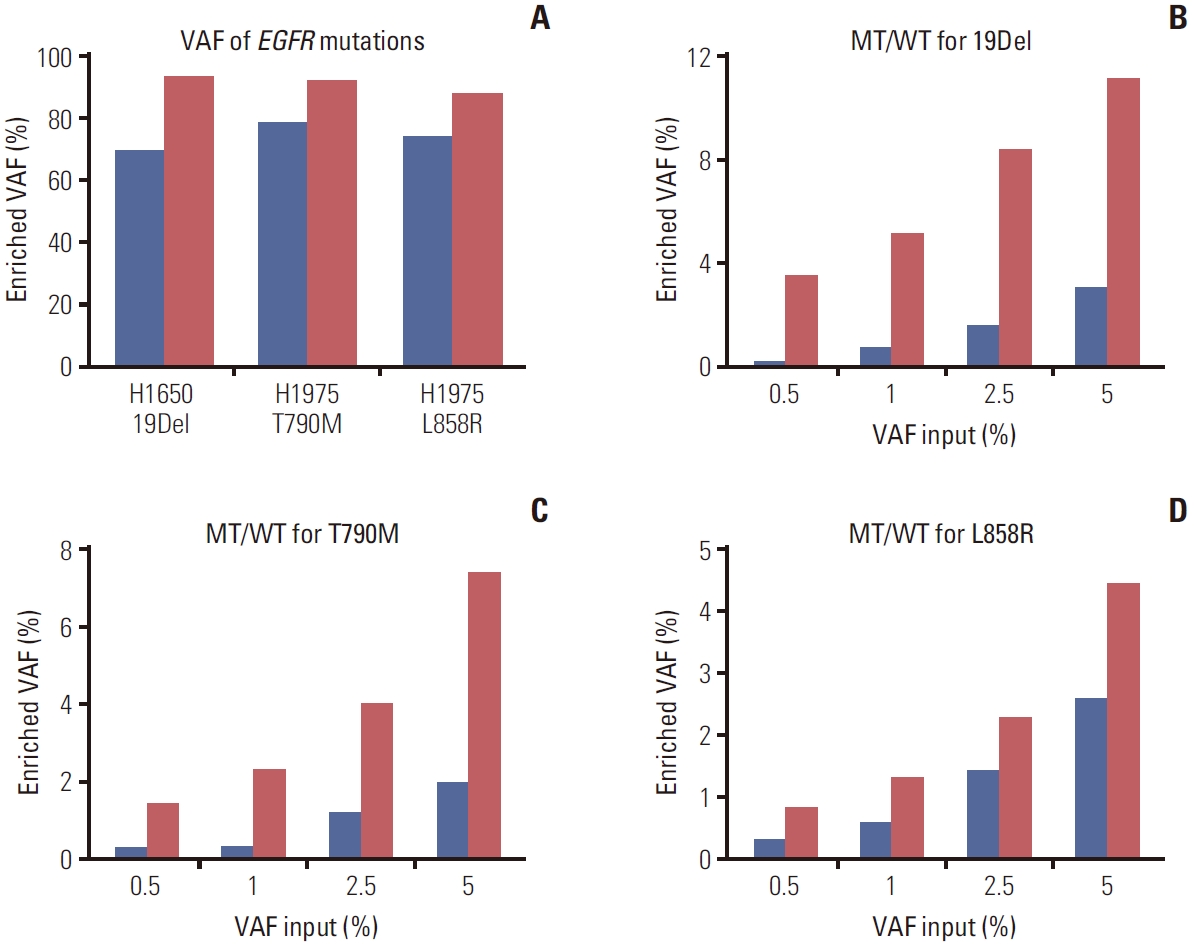
To develop a new technique for detecting EGFR mutations, BDA-based PCR amplicon Nanopore sequencing was designed and tested in lung cancer cell lines (H1650 and H1975) harboring EGFR mutations. These cell lines were used as the template in the PCR reaction with and without blocker to compare VAF enrichment. The amplified amplicons of exon 19, 20, and 21 (S4 Fig.) were sequenced by Nanopore sequencing. The result showed that the technique could increase VAF ranging from 93.38%, 92.10%, and 87.98% of EGFR 19Del, T790M, and L858R mutations, respectively, which were more than the average of 16.79% of the samples enriched by without blocker (Fig. 2A, S5 Table). Furthermore, all nucleotide mutations detected by the assay could be viewed on IGV, with a few sample instances presented in S6 Fig.
(2) Evaluation of the assay for detecting low VAF
To evaluate the assay for detecting low VAF in clinical specimens, the mutant samples input with different ratios of 5%, 2.5%, 1%, and 0.5% in a total DNA sample of 106 copies were sequenced. The results indicated that BDA assay led to higher VAF enrichment than without blocker, the average of 6.80, 4.69, and 2.04-fold for exons 19, 20, and 21, respectively (Fig. 2B-D). Moreover, the detection limit was established at the lowest ratio of 0.5% enriched to 3.57%, 1.45%, and 0.83% for the mutations in exons 19, 20, and 21, respectively. However, the assay for detecting 0% mutant EGFR was found to have background noise from PCR amplification errors and a base-calling accuracy of 96.84% based on a Q score ≥ 15 (S7 Table).
2. Validating in clinical tissue specimens
(1) Calculated cutoff for detection
To distinguish between the sequencing error and low VAF, we calculated cutoff for detection following equations 1 and 2. Each EGFR mutant was serial diluted from 5% to 0.1% VAF and sequenced. The result found that exon 19 showed a mean enriched VAF of 5.81% (range, 0.73% to 11.13%), for exon 20 showed a mean enriched VAF of 2.99% (range, 0.09% to 7.04%), and for exon 21 showed a mean enriched VAF of 1.79% (range, 0.06% to 4.45%). The calculated sequencing error for wild-type samples indicated values of 1.22%, 0.49%, and 0.29% of exon 19, 20, and 21, respectively. Thus, the calculated cutoff for this technique was 3.09%, 1.44%, and 0.74% for 19Del, T790M, and L858R, respectively (Fig. 3A, C, and E).
(2) Evaluation of VAF in EGFR mutations
BDA amplicon Nanopore sequencing was applied for detecting EGFR mutations in FFPE DNA samples. The results showed that the VAF for 19Del ranged from 13.47% to 93.33%, T790M ranged from 6.41% to 49.46%, and L858R ranged from 17.10% to 81.78%, as shown in S8A Fig. In addition, 35 of 65 samples from NSCLC patients were detected with EGFR mutations, including 19Del (12 patients), L858R (13 patients), and double mutations as 19Del, T790M (5 patients) and T790M, L858R (5 patients). These results were in concordance with detection by the standard method based on Cobas real-time PCR, as shown in Table 2.
3. Validating in clinical blood specimens
(1) Calculated cutoff for detection
The validation of the technique for detecting EGFR mutations in cfDNA samples revealed the assay failed to amplify the EGFR gene using the same approach as for FFPE (data not shown). Subsequently, we optimized the BDA method to amplify EGFR mutations from cfDNA (S9 Table for the method). To determine a cutoff for detecting low VAF, wildtype cfDNA samples were used to calculate the VAF from sequencing error. The results found that the VAF was 3.06%, 0.23%, and 0.21% for exon 19, 20, and 21, respectively. The calculated cutoff was 4.94% for 19Del, 1.18% for T790M, and 0.66% for L858R mutations (Fig. 3B, D, and F).
(2) Evaluation of VAF in EGFR mutations
Thirty-eight of 50 samples were successfully amplified and sequenced. The VAF assessment found that the assay could enrich 19Del in a range from 13.25% to 66%, T790M in a range from 3.71% to 41.39%, and L858R in a range from 4.70% to 70.74% (S8B Fig.). Moreover, 29 of 38 cfDNA samples were detected EGFR mutations, including 19Del (15 patients), L858R (3 patients) and double mutations as 19Del and T790M (4 patients), T790M and L858R (2 patients), 19Del and L858R (5 patients). The result contained 12 false-positive samples for 19Del and one sample for L858R. Subsequently, these false-positive samples were validated through Barcode Taq (BT) sequencing. The results confirmed 10 of 12 cfDNA samples as true-positive for 19Del and one sample as truepositive for the L858R mutation. These results were consistent with the outcomes obtained from the ddPCR-based detection method, as shown in Table 3.
Discussion
In this study, we developed the technique to enable low VAF from EGFR mutations detection by BDA amplicon Nanopore sequencing for cost-effective detection. The assay demonstrated that using BDA significantly increased VAF. Despite mutant EGFR is normally presented at a very low level in NSCLC patients, this technique achieves detection of at least 0.5% VAF, compared to the NGS identify at least 5% VAF [19].
Furthermore, the assay was validated to detect EGFR mutations in clinical samples. Using multiple PCR cycles and more blocker concentration may lead to error detection of 19Del in cfDNA [20,21]. These were observed from calculated VAF from sequencing error of wild-type patients, resulting in the cutoff higher than detecting EGFR mutations in FFPE.
For the testing with FFPE DNA, one sample was identified as a false-positive for the 19Del mutation. Subsequently, this sample was confirmed through real-time PCR, revealing it to be wild-type EGFR. Consequently, the detection of 19Del had a high concordance rate of 98.46% and 100% for T790M and L858R mutations.
For testing with cfDNA samples, 12 of 50 samples failed to amplify and were subsequently excluded, which might have low levels of cfDNA in the blood or degraded DNA. The assay found that T790M and L858R were 100% consistent with the ddPCR. Moreover, two samples were false-positive for 19Del mutation, resulting in a concordance rate of 94.74%. However, there are a few possible reasons for the discordance rate. Firstly, some NSCLC patients had EGFR exon 19 deletion of 10 bases (2243_2252_10del), hence Cobas EGFR probe for 19Del (15 bases deletion) would fail to capture. Secondly, some patients actually have rare double L858R and 19Del mutations [21,22]. According to this finding, 19Del and L858R mutations can potentially modify the molecular conformation of the EGFR-TK ATP binding site, particularly in the active domain [21,23]. This alteration may lead to reduced binding of EGFR-TKIs to the EGFR-TK domain, ultimately affecting the response to EGFR-TKIs [24]. However, the potential impact of concurrent mutations in exon 19 and 21 on clinical outcomes remains unclear [22]. Lastly, the BDA could enrich low VAF, suggesting that the assay might be more sensitive than the standard technique [14].
In the raw reads for sequencing, after base-calling using a Q score ≥ 15 results in a loss of approximately 36.21% of the total reads. However, a coverage depth of 1,000 reads is sufficient to detect gene mutation. Thus, for detecting three regions within one sample, a minimum raw read of 5,000 and reads per sample for FFPE DNA, and 7,500 reads per sample for cfDNA are recommended.
This technique could identify additional coincidental mutations within the range of amplicons, compared with the standard method that can only detect known mutations using specifically designed primers and probes. Moreover, the approach is portable and more affordable compared to the second-generation sequencers, permitting its usage in the low resource-setting areas. However, the turn-around time for the Cobas real-time PCR or ddPCR method was roughly 2 hours, significantly shorter than the turn-around time of this technique (approximately 3 days).
Nevertheless, this method had some limitations. First, the method is developed for detecting only common EGFR mutations (19del, T790M, and L858R), which does not cover other rare EGFR mutations (S10 Table). Second, this study used the SQK-LSK114 kit for long-read sequencing. However, ONT plans to release a sequencing kit for short fragments, which might be appropriate for sequencing the DNA extracted from FFPE and cfDNA.
In conclusion, the development of BDA Nanopore sequencing proves to be highly sensitive for detecting EGFR 19Del, T790M, and L858R mutations in NSCLC and could be applied in clinical settings.