Introduction
Almost 2.2 million cases of lung cancer are diagnosed annually worldwide, with non–small cell lung cancer (NSCLC) being the most common histologic type of lung cancer, accounting for 82% of all diagnosed cases [1]. Overall, 10% of patients with NSCLC in Western countries and 35% in Asia carry epidermal growth factor receptor (EGFR) mutations, which are predictive markers of response to EGFR tyrosine kinase inhibitors (TKIs) [2]. EGFR-TKIs have significantly improved clinical outcomes in advanced EGFR-mutant (EGFRm) NSCLC [3,4].
For patients with operable EGFRm NSCLC, surgery is still the backbone of treatment. Although surgical resection can achieve local control, the rates of loco-regional recurrence and systemic metastasis remain very high. Adjuvant EGFR-TKI therapy significantly improves disease-free survival (DFS), with a 3-year DFS rate of 34%–80% in EGFR-TKI groups versus 20%–28% in control groups [5,6]. Early preoperative systemic administrations of EGFR-TKIs may reduce disease stage, facilitate surgical resection, and reveal drug sensitivity to guide postoperative treatment. Therefore, the unmet need for neoadjuvant EGFR-TKI therapy is driving the exploration of its potential to improve prognosis [7].
The peculiar characteristics of the tumor microenvironment (TME) in EGFRm NSCLC may modulate the antitumor immune response [8]. According to preclinical studies, EGFR activation can increase intrinsic programmed death-ligand 1 (PD-L1) expression in tumor cells, which causes T-cell apoptosis and contributes to immune escape in EGFRm NSCLC [9]. Additionally, EGFR-TKIs can increase T cell–mediated tumor death by potentiating MHC class I and II molecule activation in response to interferon γ [10,11]. Although TME changes induced by EGFR-TKI therapy could determine the treatment efficacy [12], very few studies have focused on such changes in the preclinical models. Herein, we not only report the clinical results of a phase II trial of neoadjuvant/adjuvant erlotinib therapy among patients with stage II/IIIA EGFRm NSCLC but also analyze the TME changes induced by erlotinib.
Materials and Methods
1. Study design
This was a single-center, single-arm phase II trial of neoadjuvant/adjuvant erlotinib therapy in patients with stage II/IIIA EGFRm NSCLCs. The trial was approved by the institutional ethics board (IRB No. NCCCTS1156) and was conducted in accordance with the ethical principles of the Declaration of Helsinki. This study is registered at clinicaltrials.gov (NCT01470716), and all patients provided written informed consent. The primary endpoint was the objective response rate (ORR) by Response Evaluation Criteria in Solid Tumors (RECIST). The study was designed by Simon’s two-stage clinical trial to test the null hypothesis that the probability of a response (complete or partial) was ≤ 25% versus the alternative that the probability was ≥ 55%. There was a 5% probability of type I error (false-positive) and a 10% probability of type II error (false-negative). The trial would terminate after the first stage if only two or fewer of the eight patients would have responded to the treatment. The trial would be rejected after the second stage if only nine or fewer of 24 total patients would respond to the treatment. Assuming a dropout rate of 10%, a total of 26 patients will be enrolled. The main inclusion criteria were an age of 18 years or older, previously untreated, potentially resectable stage II or IIIA (by American Joint Committee on Cancer 7th edition TNM staging system) EGFRm NSCLC, Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, and adequate organ function. The main criteria for exclusion included a history of malignancies other than lung cancer during the preceding 3 years, any prior systemic anticancer therapy, and any unstable systemic diseases.
All patients included in the trial received neoadjuvant erlotinib therapy (NE) (150 mg/day) for 4 weeks and tumor response was evaluated (see the tumor and patient response assessment section). Tumor responses were defined as follows: complete response (CR; the disappearance of all target lesions), partial response (PR; ≥ 30.0% reduction in the sum of the diameters of the target lesions), progressive disease (PD; ≥ 20.0% increase in the sum of the diameters of the target lesions), and stable disease (SD; do not qualify as PR or PD). CR, PR, and SD with decreasing tumor patients received an additional 4 weeks of NE before surgery. Patients who continued showing CR and PR at post-surgery received adjuvant erlotinib postoperatively for 4 months otherwise patients showing SD/PD at post-surgery received 4 cycles of vinorelbine and cisplatin adjuvant chemotherapy (VP); vinorelbine (25 mg/m2 intravenously on days 1 and 8) and cisplatin (60 mg/m2 intravenously on day 1) every 3 weeks. PD and SD of increasing tumor patients after 4 weeks of NE were directly sent for surgery and received 4 cycles of VP adjuvant therapy. Further, TME response to NE were analyzed using pretreatment biopsy and resected tumor samples.
2. Tumor and patient response assessment
Patients took chest and abdomen computed tomography (CT) scans, brain magnetic resonance imaging (MRI) or CT, and positron emission tomography (PET) at baseline (pretreatment). Assessments were performed after 4 weeks of NE (additional assessments after 8 weeks for patients undergoing 8 weeks of NE) as well as 4 weeks after adjuvant therapy and every 3 months thereafter during long-term follow-up. Clinical response to treatment was evaluated using MRI, CT, and PET images according to the RECIST ver. 1.1. The ORR is the proportion of patients with CR or PR, while the disease control rate (DCR) is the proportion of patients with CR, PR, or SD. DFS is the time from assignment to the study until the date of disease recurrence or death from any cause, whichever occurred first. Overall survival (OS) is the time from the start of the study to the date of death or the last follow-up for live patients at the study end. Data were censored for patients who were alive at the last follow-up date. Adverse events (AEs) were collected according to the National Cancer Institute Common Terminology Criteria for Adverse Events (ver. 5.0).
3. Next generation sequencing analysis
DNA from formalin-fixed, paraffin embedded (FFPE) pretreatment/baseline (BL) and resected (OP) tumor tissues were extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA), according to the manufacturer’s instructions. Initial quality control (QC) checks of FFPE DNA were performed using electrophoresis on 1% agarose gels and the Qubit dsDNA HS Assay Kit with the Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA) according to manufacturer’s instructions. FFPE DNA was sheared into fragments of mean peak size of ~180–200 base pairs using Adaptive Focused Acoustics (Covaris, Woburn, MA). Libraries were prepared with the SureSelect XT protocol (Agilent Technologies, Santa Clara, CA) with Axen Cancer Master (559 genes) developed by Macrogen (Seoul, Korea). DNA fragment quality was checked using the 2100 Bioanalyzer (Agilent Technologies). A product size of ~200–400 base pairs was required. Then, the libraries were quantified using the Qubit dsDNA HS Assay Kit and the Qubit 2.0 fluorometer (Life Technologies). The libraries were sequenced paired end (2×150 base pairs) on a NextSeq500 instrument (Illumina, San Diego, CA) with high output using Sequencing by Synthesis chemistry to a depth of approximately 2,000× coverage. The adapter sequences were removed by cutadapt. Trimmed reads were aligned to the reference genome (GRCh37/hg19) using BWA-MEM. Poorly mapped reads with mapping quality below 20 were removed using Samtools ver. 1.3.1. Duplicated reads were removed with Picard MarkDuplicates (ver. 2.2.4). The base quality of deduplicated reads was recalibrated using GATK BaseRecalibrator. Somatic mutations including single nucleotide variants (SNVs) and small insertions and deletions (INDELs) were identified using the MuTect2 algorithm. The list of the SNVs, INDELs, and fusions is in S1 Table. Then, false positive variant calls originating from oxoG artifacts were excluded. Also, mutations with less than 2% variant allele frequency and 100× total depth were excluded. Germline variants were excluded when minor allele frequency (MAF) ≥ 5% in ExAC_EAS or MAF ≥ 5% in the Macrogen Korean Population Database. All remaining variants were annotated using SnpEff & SnpSift v4.3i with dbNSFP v2.9.3. The microsatellite instability (MSI) was calculated as the ratio of unstable sites over QC passed predefined regions of MSI-related sites using mSINGS. The tumor mutation burden is reported as the number of mutations per megabase (mut/Mb) of the passed missense mutations.
4. NanoString gene expression analysis
Gene expression profiles were assessed using the Nano-String PanCancer and Immunology panel using DNA from FFPE tumor tissues. DNA quality was assessed using Nano-StringQCPro. The R NanoStringNorm package was used to perform quantile normalization procedures by adjusting background correction. In total, gene expression profiles were acquired for 1,377 genes, excluding housekeeping genes in the two panels. After preprocessing, we used the t test method to identify differentially expressed genes (DEGs) in BL and OP tumor tissues. Additionally, we identified DEGs between PR and SD/PD patients using OP tumor tissues. Gene set enrichment analysis (GSEA) for DEG sets was performed using a chi-squared test to reference pathways provided in NanoString gene profile panel. Survival analysis was performed for every gene in the gene expression groups exhibiting high (> 50%) or low (< 50%) expressions in BL samples. Survival analysis was performed using SurvFit and SurvDiff functions of survival package in R (R Foundation for Statistical Computing, Vienna, Austria).
5. Statistical methods
The safety analysis population included all patients who received at least one dose of erlotinib. Efficacy analyses were based on the intention to treat population. Standard statistical methods, including descriptive statistics, χ2 test, Kaplan-Meier method, stratified log-rank test, and Cox regression model were used. Data analysis was performed using SPSS ver. 25.0 statistical software (IBM Corp., Armonk, NY) and GraphPad Prism ver. 8.0 (GraphPad Software, San Diego, CA).
Results
1. Study flow and patients characteristics
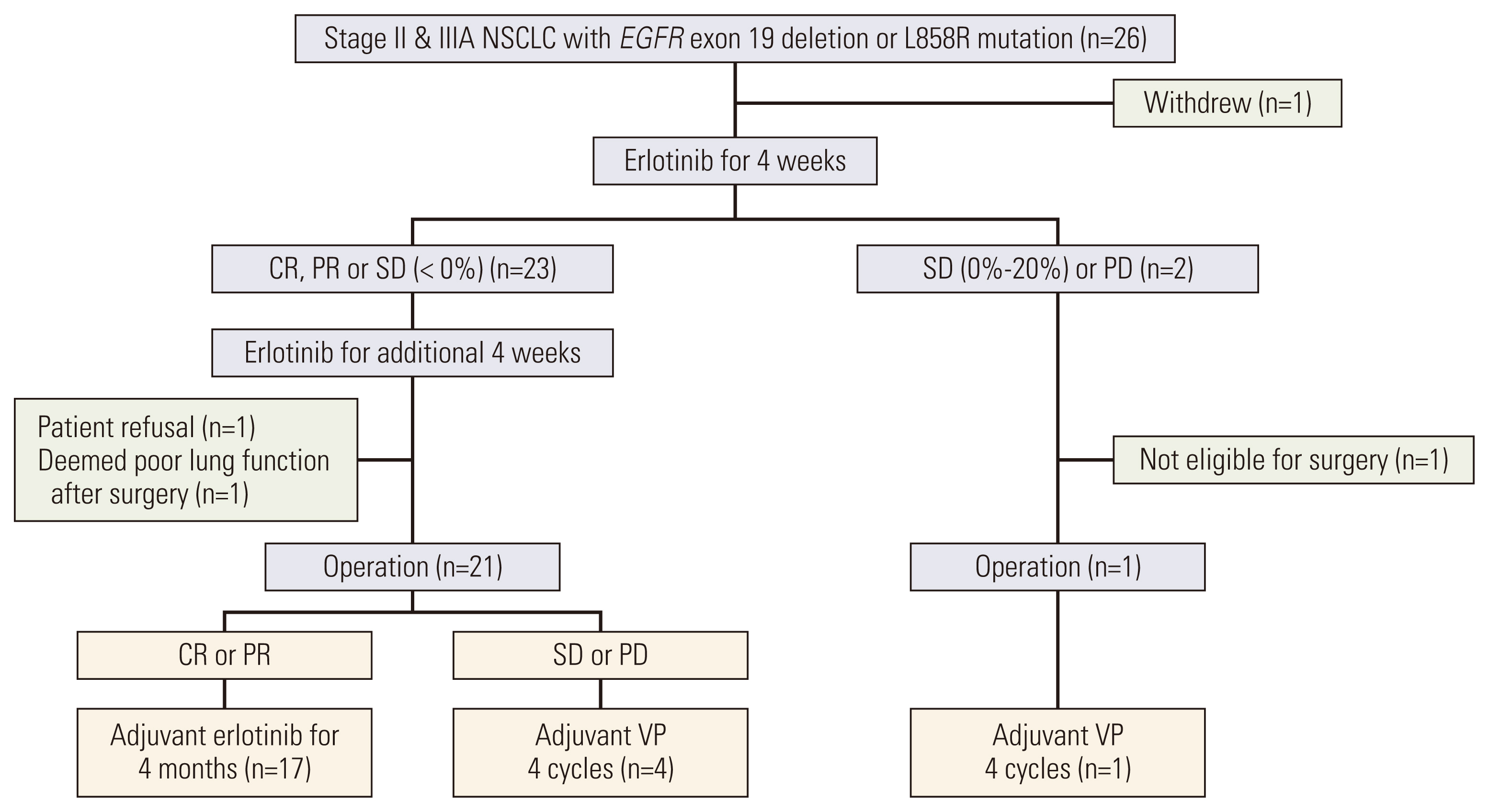
From December 2012 to November 2019, 26 patients were enrolled, however, one patient refused to participate in the clinical trial and withdrew informed consent immediately. Thus, the efficacy and safety analysis was performed for 25 patients. The total disposition of the patients is shown in Fig. 1.
Twenty-three patients achieved a 0% or more reduction in the sum of the diameters of target lesions after 4 weeks of NE and, therefore, received an additional 4 weeks of NE. After 8 weeks of NE, two patients did not undergo surgery; one patient refused to undergo surgery, and another was deemed unfit for surgery. Out of the 21 patients who underwent surgery, 16 patients continued to exhibit CR/PR and were administered 4 months of adjuvant erlotinib therapy. The remaining five patients, who exhibited SD/PD after surgery, received 4 cycles of VP.
On the other hand, two patients showed SD with increasing tumor size or PD after the initial 4 weeks of NE. Out of these two patients, the patient showing PD underwent lobectomy and received 4 cycles of VP.
Patient clinical characteristics are listed in Table 1. The median age was 61 years; 69% of patients were female. A majority of the patients had adenocarcinoma histology (85%) and had never smoked (73%). Of the total patients, 62% harbored an EGFR exon 19 deletion and 38% an exon 21 substitution (L858R) mutation. A majority of the patients were stage IIIA (88%), and the remaining patients (n=3, 12%) were stage IIB.
2. Treatment outcomes
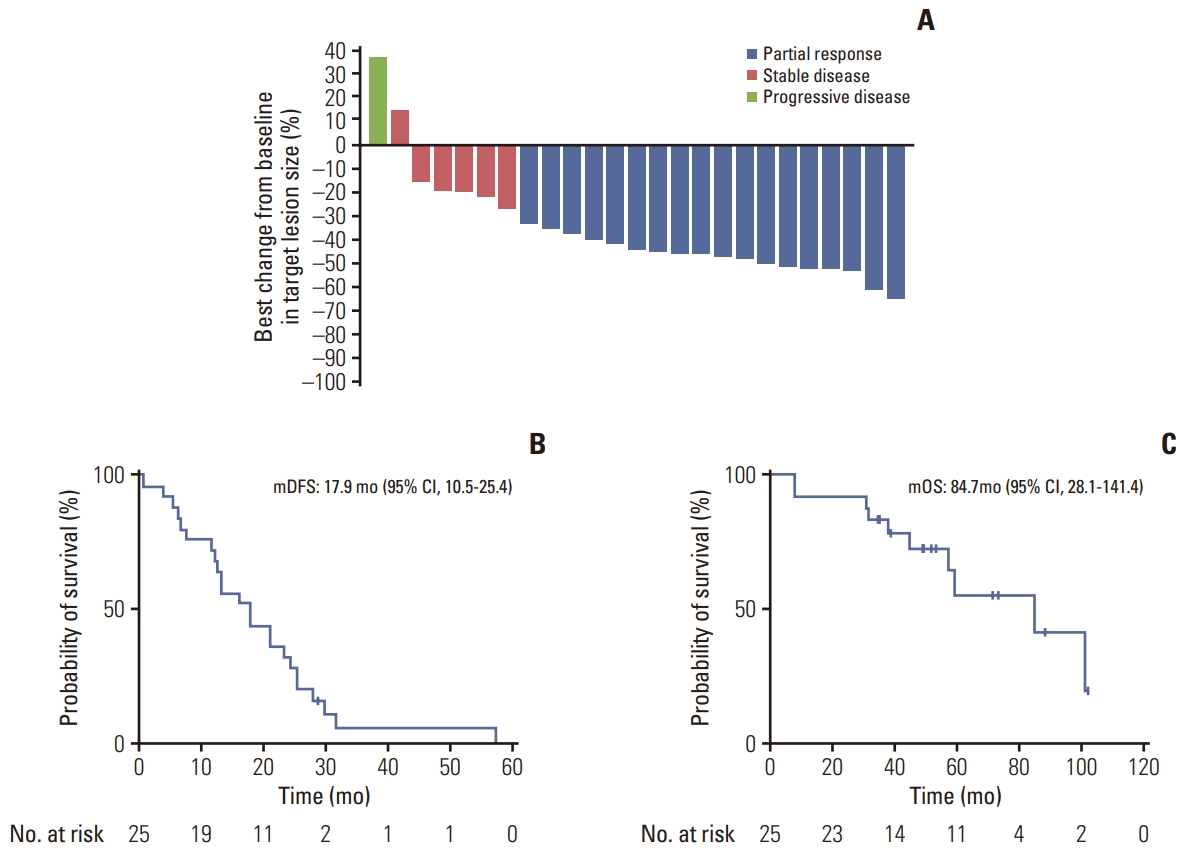
The ORR and DCR for NE were 72% (95% confidence interval [CI], 52.4 to 85.7) and 96% (95% CI, 80.5 to 99.3), respectively. The median best percentage change in target lesion size was –45% (range, –64.7% to 37%). A waterfall plot that highlights the degree to which each patient responded to erlotinib is shown in Fig. 2A.
Postoperative outcomes are listed in Table 2. After NE, 22 patients underwent surgery. Nineteen patients received lobectomy, two patients received bilobectomy and one received pneumonectomy. In addition to the clinical responses, the pathologic responses were assessed by histologic analysis of the OP tissues to recognize any remaining malignant cells, providing a more accurate assessment of the NE efficacy. All patients had R0 resections and there were two cases where N2 downstaging occurred. Pathologic complete responses were absent.
As of April 2022, the median follow-up duration was 52.8 months (range, 7.7 to 101.8 months). The median DFS and OS were 17.9 months (95% CI, 10.5 to 25.4) and 84.7 months (95% CI, 28.1 to 141.4), respectively (Fig. 2B and C).
3. Adverse events
All the patients developed at least one adverse event; four (15%) experienced grade 3 or higher toxicities from neoadjuvant/adjuvant therapy. Grade 3/4 toxicities that occurred were diarrhea (4%), elevated serum aminotransferase (4%), dyspnea (4%), and elevated bilirubin (4%). Grade 5 events were not observed. S2 Table lists AEs associated with NE. Rash (96%), diarrhea (60%), cough (60%), and pruritus (52%) were the most common AEs. One patient with grade 3 diarrhea had dose reduction and five patients had temporal discontinuation of erlotinib mainly due to aminotransferase or bilirubin increment.
4. Postoperative recurrence and subsequent treatment
Among patients (n=21) who experienced a recurrence after surgery, 12 underwent rebiopsy. Eight patients retained the baseline EGFR mutation in their tumors and one patient obtained extra exon 20 T790M resistance mutation after recurrence. Tumors in two patients lost their EGFR-TKI sensitizing EGFR mutation. EGFR mutation testing was not performed in two patients. Three patients experienced loco-regional recurrence, while 18 patients had distant relapse. Among the cases of distant relapse, nine patients had lesions in the lung, three had lesions in the brain, and one had a lesion in the pleura as a single relapsed site. Additionally, three patients had multiple relapsed sites; one patient had lesions in the brain and lung, while the other two had lesions in the lung, bone, and liver. Of the patients who experienced recurrence, subsequent therapy consisting of a first-generation EGFR-TKI for 15 (71%), a second-generation EGFR-TKI for two (10%), platinum-based chemotherapy for two (10%), and single-agent chemotherapy for one (5%) was administered (thus, overall, 20 [95%] received subsequent therapy).
5. Gene expression profile and its correlation with response and survival
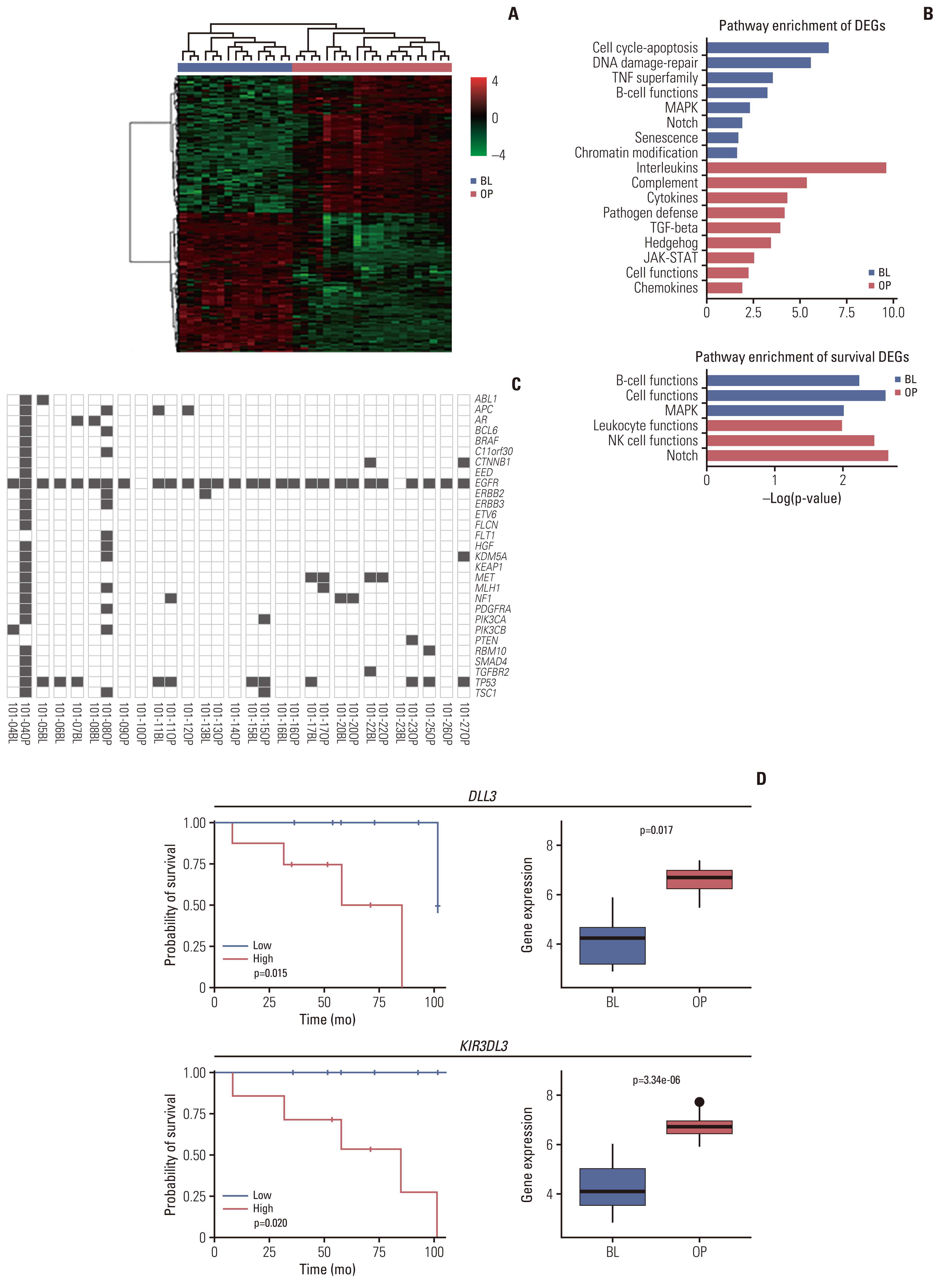
Pathogenic driver mutations for lung cancer were identified using somatic mutation profiles extracted from 31 patient tissue samples using next generation sequencing (Fig. 3C). Next, 357 and 349 DEGs upregulated in BL and OP tissues, respectively, were identified (p < 0.001) (Fig. 3A and B). For each upregulated gene, enrichment pathways were acquired using GSEA (S3–S5 Tables). Genes involved in DNA damage repair, cell cycle apoptosis, and B-cell function pathways were overexpressed in BL tissues. Meanwhile, those involved in interleukin, complement, cytokine, transforming growth factor β (TGF-β), and hedgehog pathways were activated in OP tissues (p < 0.05). To identify predictors of survival, we interrogated OS significance for DEG expression. The B-cell functions and mitogen-activated protein kinase pathways were enriched in BL tissues and Notch, natural killer cell, and leukocyte function pathways were enriched in OP tissues. The survival curves for selected DEGs from the high and low gene expression groups are shown in S6 Fig. Especially, DLL3 overexpression in BL tissues was associated with poor outcomes (p=0.015), and DLL3 expression level increased in OP tissues (p=0.017). High expression of KIR3DL3 in BL tissues was also associated with poor outcomes (p=0.020) and the association was stronger for KIR3-DL3 expression levels in OP samples (p < 0.001) (Fig. 3D). While the expression of hallmark immune genes CD8 and TIM3 was increased, that of PDL1 and LAG3 decreased in OP tissues. However, the expression level of these genes was not significantly associated with survival (S7 Fig.). Pathogen defense, interleukin, and T-cell function pathways gene sets were upregulated in the OP tissue of the PR group (n=16, p < 0.2). While, in SD/PD group (n=6), cell cycle pathway genes (ATR and CDK4) were over-expressed and significantly correlated with survival (Fig. 4, S8 Fig.)
Discussion
The existing standard of care adjuvant cisplatin doublet and neoadjuvant cisplatin-based chemotherapy show similar degrees of benefits in operable early-stage lung cancer; a pooled meta-analysis demonstrated a 5% improvement in the 5-year OS [13,14]. However, incorporation of biomarker-matched targeted therapies such as EGFR-TKIs into the adjuvant and neoadjuvant settings have been delayed by (1) the difficulty of routine predictive biomarker testing in early-stage lung cancer and (2) the long duration of trials, historically designed with DFS as the primary endpoint.
Randomized phase II and subsequent phase III trials on adjuvant EGFR-TKI therapy conducted in China compared adjuvant gefitinib with cisplatin doublet chemotherapy in patients with resected stage III-N2 EGFRm NSCLC. Improvements in DFS, which was the primary endpoint of these trials, was reported; however, OS was not affected [5,6]. In 2020, the phase III ADAURA study led to the approval of the use of third-generation EGFR-TKI osimertinib for adjuvant therapy (≤ 3 years duration). Osimertinib improved DFS with a hazard ratio of 0.20 (p < 0.001) compared to that by placebo [15]. Neoadjuvant EGFR-TKIs can also achieve clinically meaningful responses and improve OS. Two, single-center phase II trials on NE efficacy in treating stage IIIA NSCLC exhibited ~40%–60% ORRs with 67% major pathologic response (MPR) rates [16,17]. Furthermore, a single-arm phase II trial of neoadjuvant gefitinib therapy for treating stage II–IIIA EGFRm NSCLC reported an ORR and MPR rate of 55% and 24%, respectively [18]. In a multicenter, randomized, phase II EMERGING-CTONG 1103 study, numerical improvement in ORR and significant improvement in DFS were observed with NE compared to other neoadjuvant chemotherapy in patients with stage IIIA-N2 EGFRm NSCLC [7]. Currently, a global phase III study named the NeoADAURA, comparing the efficacy of osimertinib, chemotherapy, and osimertinib+chemotherapy combination in the neoadjuvant therapy, is ongoing (NCT04351555).
Here, in addition to investigating the NE benefits in treating stage II/IIIA EGFRm NSCLC, we studied its effects on TME. Gene expression analysis of OP specimens revealed that NE upregulated the immune pathways related to interleukins, complements, and cytokines. Contrastingly, NE upregulated the hallmark immunosuppressive pathways of TGF-β and hedgehog. TGF-β crucially promotes cancer progression by activating cancer-associated fibroblasts, stimulating angiogenesis, and suppressing the immune system [19]. During continuous erlotinib treatment, these unfavorably upregulated immune suppressive pathways may promote resistance development. TGF-β, the critical TME regulator, is secreted by multiple types of stromal and tumor cells. TGF-β signaling activated by TKI therapy increases the expression of EGFR, PDFGR, ERK, AKT/STAT which activate alternative survival pathways which cause resistance to TKIs [20]. The hedgehog pathway modulates stromal cells to create an immunosuppressive TME, which fosters cancer progression by driving M2 polarization of tumor-associated macrophages [21]. Additionally, aberrant activation of the hedgehog signaling pathway is implicated in the development of EGFR-TKI resistance through the induction of the epithelial-to-mesenchymal transition [22]. Further trajectory analysis is warranted since even if immune-mediated anticancer effects of EGFR-TKIs are temporary, certain immunosuppressive factors may accumulate gradually over the prolonged course of treatment. Further, the expression of the well-known individual immune checkpoint genes, PDL1 and LAG3 decreased in OP samples. Concurrently, anti–programmed death-1 response in EGFRm NSCLC after the development of TKI resistance is abysmally low [23]. Moreover, EGFR-TKI therapy increases the expression of the immune checkpoint gene TIM3, indicating that pharmaceutical inhibition of TIM3 can overcome resistance.
Notably, gene sets upregulated in OP samples varied with tumor response to NE. In the PR tumors, immune pathway genes were upregulated. Meanwhile, in the SD/PD tumors, cell cycle-apoptosis pathway genes were upregulated. Tumor response was associated with DFS. Median DFS was 18.5 months for the PR patients and 13.2 months for the SD/PD patients. Several NSCLC mRNA expression microarray studies have identified expression signatures that stratify patients into prognostic groups [24]; however, there is discordance between these putative prognostic gene lists due to heterogeneity in patient cohorts. To the best of our knowledge, our study is the first to analyze gene expression during EGFR-TKI therapy in a restricted neoadjuvant setting cohort. From the results, we speculated that activation of certain pathways could modulate the EGFR-TKI treatment response.
DLL3 overexpression at BL was associated with poor outcomes. DLL3 is a Notch family ligand that regulates tissue homeostasis through cell renewal, differentiation, proliferation, and cell death [25]. DLL3 high expression is associated with a poor prognosis of breast, bladder, and small cell lung cancers [26]. Similar associations have not been found yet for EGFRm NSCLC. However, EGFR signaling negatively controls Notch1 expression, through a mechanism involving transcriptional downregulation of p53 [27]. Thus, continuous EGFR inhibitor therapy enriches ALDH expressing stem-like cells in a Notch3-dependent manner, causing resistance [28]. Zou et al. [29] showed that Notch1 expression is upregulated in gefitinib-resistant lung cancer and that upregulation could be reversed by Notch inhibition resulting in increased apoptosis. Our study suggests that EGFR blockade response may differ based on Notch3-dependent signaling at baseline and that Notch signal suppression and EGFR inhibition may synergize to induce apoptosis.
Recently, studies on human endogenous retrovirus-H long terminal repeat-associating 2 (HHLA2), a promising immune checkpoint protein, are flourishing because of its newly discovered co-inhibitory receptor called the killer cell Ig-like receptor, three Ig domains, and long cytoplasmic tail (KIR3DL3) [30,31]. KIR3DL3 is a killer cell immunoglobulin-like receptors (KIR) family member. It endows its ligand, HHLA2, with functions related to cancer immunity. HHLA2 expression is correlated with unfavorable prognosis across multiple human cancers [32]. KIR3DL3-HHLA2 binding may attenuate CD8+ T cells–mediated lysis, irrespective of T-cell receptor engagement, in vitro [30]. Agreeingly, we found that KIR3DL3 expression is associated with worse outcomes. Thus, we suggest, that in addition to being a prognostic marker in EGFRm NSCLC, KIR3DL3 is a potential target for enhancing antitumor immunity via blockade.
A few limitations of this trial include its (1) limited sample size; (2) single-arm nature; and (3) short duration of adjuvant erlotinib therapy (only 4 months). The currently approved EGFR-TKI adjuvant therapy involves 3 years of treatment with osimertinib. The concern of losing the opportunity for surgery after neoadjuvant treatment is also an issue. However, the outcomes (ORR, DFS, OS, and surgery rate) of this trial were comparable to those observed in previous clinical trials. Moreover, genetic analysis (for mutation and gene expression profiles) identified several pathways related to clinical outcomes. Thus, this study is insightful in identifying novel therapeutic targets (such as DLL3 and KIR3DL3 in EGFRm NSCLC) to circumvent the development of anticancer drug resistance.