Introduction
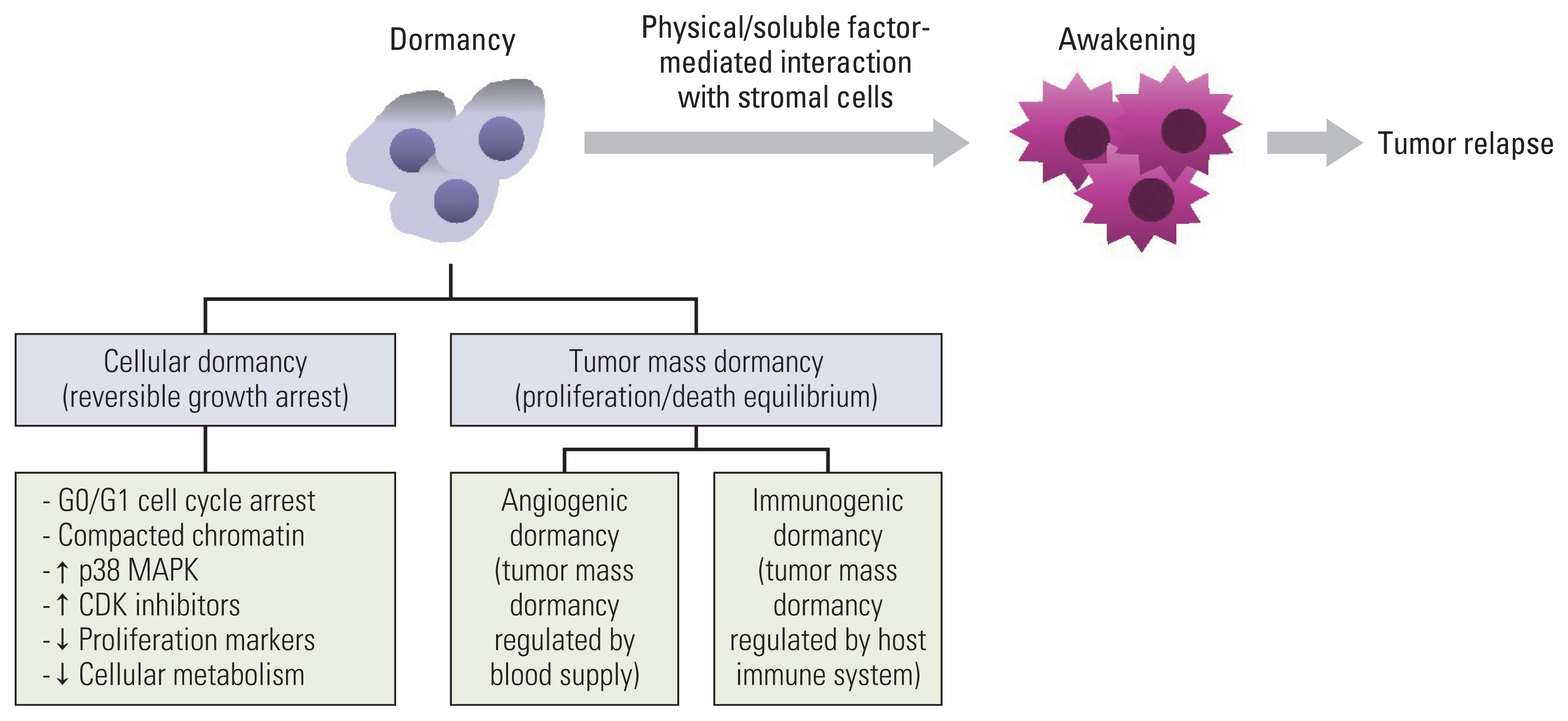
Cancer is one of the main causes of death worldwide and a great impediment to extending life expectancy [1]. Anticancer drug resistance and tumor relapse, which may even occur several years after curative anticancer therapies, are the main causes of cancer-associated mortality [2]. Recurrent tumors are believed to emerge from a subpopulation of cells that survive anticancer treatment, and are generally referred to as minimal residual disease (MRD). They may remain in a clinically undetectable state for some years or decades and then resume growth under certain growth-favoring circumstances to develop recurrent tumors [3,4]. MRD covers the following populations: a rare subpopulation of cancer cells with resistance to anticancer therapeutics in therapy-sensitive tumors; disseminated tumor cells (DTCs) that escape from the primary tumor, survive against environmental stresses and the immune system in circulation, and arrive at and colonize distant organs in the early or late stages of cancer progression; and circulating tumor cells that move in the bloodstream or lymphatic system and finally become DTCs [4–7]. Although adjuvant therapy administered in certain cases is deemed to suppress tumor relapse by targeting the MRD, not all patients benefit from it. Therefore, MRD is considered a marker of poor prognosis in patients with cancer, and understanding the mechanisms by which tumors remain dormant or awake is fundamental for developing strategies to prevent tumor relapse. Indeed, owing to substantial progress in deciphering the mechanisms underlying cellular dormancy and awakening during the last decades, several studies have reported on the biology of cellular dormancy and potential targeting strategies [8–12]. Hence, this review article aims to provide mechanistic insights into cancer cell dormancy and awakening, discuss the implications of cellular dormancy in chemoresistance and cancer progression, and propose potential strategies to develop dormancy-targeting anticancer agents. A brief summary of the mechanisms responsible for maintenance of and escape from dormancy is presented in Fig. 1.
Tumor and Cellular Dormancy
Tumor dormancy is a critical phase in cancer development during which tumor cells exist, but tumor progression is not clinically apparent [13]. It includes both tumor mass dormancy, referring to the presence of neoplastic masses that have achieved a balance between cell proliferation and apoptosis [8,9,14], and cellular dormancy, referring to a slow-cycling or non-proliferative state with reversible and temporal growth arrest. As described before, tumor mass dormancy is the equilibrium status between cell proliferation and cell death [8,9,15], which is further subclassified as “angiogenic dormancy” and “immunogenic dormancy” [8,9]. Angiogenic dormancy is a phenomenon that occurs prior to the angiogenic switch, in which cell proliferation and death are balanced based on the level of oxygen and nutrients supplied by the preexisting blood vessels [8,9,15]. Before the angiogenic switch, increased cell proliferation causes depletion of oxygen and nutrients in the environment distal to the blood vessel, eventually leading to cell death and an equilibrium between proliferation and the death of cancer cells [8,9,15]. Supplementation of angiogenic factors awakened dormant tumor cells and promoted tumor growth [9,16]. Immunogenic dormancy is the status of balanced cell proliferation and cell death based on surveillance of the host immune system [8,9]. Cancer cell proliferation and tumor growth can be suppressed by surveillance of the host immune system, including T cells, B cells, and natural killer (NK) cells that recognize and kill tumor cells through tumor-specific or stress-induced cell surface antigens [17]. In addition, interferon-γ (IFN-γ), produced by Th1 cells, CD8+ T cells, NK cells, and NK T cells, induces dormancy in cancer cells via signal transducer and activator of transcription 1 (STAT1)–dependent or –independent pathways (for example, the indolamine 2,3-dioxygenase 1 [IDO1]–kynurenine [Kyn]–aryl hydrocarbon receptor [AhR]–p27 pathway) [18–22]. If the host immune system cannot completely eliminate cancer cells or maintain their dormant status, the remaining cancer cells may proliferate; however, the expansion of cancer cells can be controlled by the host immune system. Therefore, an active host immune system dynamically balances the proliferation and death of cancer cells, causing immunogenic dormancy [8,9,17]. The evolution of dormant cancer cells to acquire the ability to blunt antitumor immunity causes the escape of the host immune system and the consequent outgrowth of dormant cancer cells [8,9,17]. In contrast, cellular dormancy is a state in which cells undergo quiescence, which is more consistent with the classical definition of dormancy than tumor mass dormancy [8,9]. Considering the shared features between dormant cancer cells and slow-cycling cancer cells (SCCs), such as therapy resistance, reversible cell cycle arrest, and a potential cause of tumor relapse [23,24], SCCs are also regarded as dormant or quiescent cells [23].
Identification of Dormant Cancer Cells and Their Characteristics
1. Identification of dormant cancer cells
To identify clinically relevant dormancy-associated biomarkers, it is necessary to establish experimental models of dormant cancer cells using clinical samples. Several approaches have been proposed for identifying dormant cancer cells in preclinical and clinical models. The existence of dormant cancer cells has been determined by evaluating the level of proliferation and apoptosis-related marker expression (for example, Ki67 and terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL] expression [Ki67 and TUNEL-double-negative population is considered as the dormant population] [25]; Ki67 and M30 expression [Ki67 and M30-double negative population is considered as the dormant population] [26]), and the mitotic activity index (the total number of mitoses in 10 fields of vision according to the Multicenter Mammary Carcinoma Project protocol) [27] or mitotic index (determination of the level of phosphorylated histone H3 expression)] [28]. Several experimental approaches, such as the pulse and chase experiment using nucleotides that can be incorporated into DNA during DNA replication [for example, BrdU (5-bromo-2′-deoxyuridine), EdU (5-ethynyl-2′-deoxyuridine), or tritiated thymidine (3H-T)], label-retention methods using carboxyfluorescein succinimidyl ester (CSFE), PKH26, or DiD dyes, the labeling and chase method using a doxycycline-inducible green fluorescence protein (GFP)–tagging histone H2B (H2B-GFP) reporter, use of the Fluorescent Ubiquitination-based Cell-Cycle Indicator (FUCCI) reporter, gene promoter reporters using CDKN2A (that encodes p16) or KDM5B promoters, and a cell cycle indicator (the mVenus-p27K− probe) have been used for identification of slow-cycling/dormant cancer cells [23,29–34]. Bioencapsulation in a stiff and porous 3D matrix using synthetic materials has been recently developed for isolating dormant cancer cells [35]. Furthermore, as described in recently published literature [36,37], several in vitro models for tumor dormancy and genetically engineered mouse models for the establishment of clinically relevant in vivo dormancy models have been developed. Several methods for identifying dormant cancer cells are summarized in Table 1.
2. Main characteristics of dormant cancer cells
Typical dormancy-associated cellular changes are reversible retardation or arrest of cell proliferation, which is accompanied by cell cycle arrest at the G0/G1 phase of the cell cycle, induction of cyclin-dependent kinase inhibitors, including p21 and p27, and downregulation of cell proliferation-related markers such as Ki67 and proliferating cell nuclear antigen [8,9,11]. Activation of p38 mitogen-activated protein kinase (MAPK) and downregulation of extracellular signal-regulated kinase (ERK) activation (p38High/ERKLow) have been considered prototype markers of dormant cancer cells [38,39]. However, in recent studies, ERK activation was also found in cells that underwent dormancy-like states, such as cells forced to acquire dormancy-like phenotypes by incubation onto biomaterials under serum deprivation conditions [40] or those with SCC-like phenotypes (drug-tolerant persister [DTP]) by treatment with irinotecan (CPT-11) [41]. In addition, despite growth arrest and/or retardation and environmental insults, dormant cancer cells exhibit minimal cell death and harbor resistance to anticancer therapeutics [15,23,34,43]. These cells also display reduced cellular metabolism, such as decreased energy production, protein translation, and glycolysis [44,45]. Dormant cancer cells also have a compact chromatin structure due to K20 methylation of histone H4 [24,46].
3. Association of senescence with cellular dormancy in cancer cells
Several dormant cell characteristics, such as growth arrest and therapy resistance, appear to be shared by other cellular alterations including senescence and cancer stem cells (CSCs). Despite the potential of reversible growth arrest in senescent cells, such as escape from chemotherapy-induced senescence reported in a recent study [47], senescent cells generally undergo an irreversible growth arrest [24,48,49]. In addition, senescent cells display persistent DNA damage responses, altered chromatin structure (senescence-associated heterochromatin foci), elevated β-galactosidase (β-gal) activity, and a hypersecretory ability known as the “senescence-associated secretory phenotype” (SASP) [24,48,49]. Senescence is induced in response to various stressful stimuli such as oncogene expression and chemotherapy [48]. Despite the presence of senescence-associated characteristics in dormant cancer cells [24,48] and the similarities between senescence and dormancy (such as induction by stressful stimuli, growth arrest accompanied by p21 upregulation, and p38 MAPK activation) [24,50], several studies have shown that senescence is not always involved in the maintenance of dormant cancer cells [42,51]. In addition, the features of senescence differ from those of dormancy in terms of the reversibility of cell cycle arrest (irreversible vs. reversible), metabolic activity (high vs. low), and interaction with the immune system (immune attractive vs. immune evasive) [24]. However, regardless of similarities and differences in the regulation of intrinsic signaling changes, senescence appears to be an aspect of dormancy in tumors and contributes to the regulation of dormancy and awakening of dormant cancer cells via its SASP phenotype [24,52] and by mediating the acquisition of epithelial-mesenchymal transition (EMT) and stemness phenotypes [53].
4. Association of EMT and/or CSC-like phenotypes with cellular dormancy in cancer cells
CSCs are a rare population of tumors harboring stem cell–like characteristics, such as self-renewal, symmetric or asymmetric division, differentiation capacity, therapy resistance, and tumorigenic activity [54,55]. CSCs are thought to arise from normal stem cells through the sequential acquisition of genetic or epigenetic changes, or from the dedifferentiation of progenitor cells harboring genetic mutations or non-CSCs [54,56]. CSCs are capable of generating diverse tumor cell populations and play crucial roles in tumorigenesis and cancer progression [54,57]. In addition, the EMT program is closely involved in the acquisition of stemness phenotypes [58–60]. The key cellular changes in the EMT program are the loss of polarity of epithelial cells with downregulation of epithelial markers (for example, E-cadherin) and gain of mesenchymal markers including N-cadherin, fibronectin, vimentin, Snail1, Snail2, Slug, Twist1, and zinc finger E-box binding homeobox 1 (Zeb1), with invasive phenotypes [59]. EMT-mediated transcription factors (Snail1, Snail2, Twist1, and Zeb1) were found to induce stemness markers including sex determining region Y-box 2 (Sox2), B lymphoma Mo-MLV insertion region 1 homolog (BMI1), and octamer-binding transcription factor 4 (Oct4) [61,62]. However, the EMT activator paired related homeobox 1 (Prrx1) was found to repress stemness in cancer cells [63], indicating that both EMT and mesenchymal-epithelial transition (MET) programs may contribute to the acquisition of stemness phenotype [61]. Moreover, although several features of dormant cancer cells differ from those of CSCs, including the low tumor-initiating properties of dormant cancer cells compared to the high tumorigenic activity of CSCs and growth arrest (quiescence) in dormant cancer cells compared to quiescent or proliferative phenotypes in CSCs [23], studies have shown that stemness phenotypes are associated with the regulation of dormancy in cancer cells [59,64–66], implying that CSC phenotype acquisition may play a role in the modulation of dormant cancer cells. Considering the complexity of EMT regulation and stemness [61], additional studies are necessary to clarify the precise role of EMT/MET and stemness programs in the regulation of dormancy.
Mechanisms Underlying Regulation of Cancer Cell Dormancy
Growth arrest in dormant cancer cells is a consequence of the intrinsic regulation of cell cycle machinery, modulation of cellular signaling through interaction with the extracellular matrix (ECM) and ECM-associated molecules, signal transduction in response to various stimuli from the surrounding microenvironment, and other cellular changes [9,11] (Fig. 2). The regulation of cellular dormancy by the aforementioned factors is detailed below.
1. Regulation of cellular dormancy by the Rb-E2F and DREAM complexes
Dormant cancer cells undergo cell cycle arrest in the G0/G1 phase. Cell cycle progression is tightly and cooperatively regulated by retinoblastoma (Rb)-E2F and the dimerization partner, Rb-like, E2F, and multi-vulval class B (DREAM) complexes in a transcription-dependent manner [67], and these mediators play an important role in mediating dormancy in cancer cells. During quiescence, the Rb protein blocks cell cycle entry into the S phase by repressing the transcription of genes responsible for the cell cycle machinery via direct binding to the transactivation domain of the canonical E2F transcription factors and chromatin structure remodeling through interaction with proteins responsible for chromatin remodeling (such as human Brahma [BRM] and brahma-related gene-1 [BRG1]) and histone modification [such as histone deacetylase 1 and histone lysine methyltransferase SUV39H1] [67,68]. Cyclin D1/cyclin-dependent kinase (CDK) 4/6 and cyclin E/CDK2 complexes phosphorylate Rb protein, causing the release of Rb protein with E2F and cell cycle progression [68]. In addition to Rb protein, noncanonical E2F proteins (E2F6, E2F7, and E2F8) repress the transcription of cell cycle regulators in an Rb protein-independent manner [67]. A previous study also demonstrated the association of activation threshold of the Rb-E2F network with the depth of quiescence [69].
The DREAM complex, which consists of an Rb family protein (p130/RBL2 or p107/RBL1), a repressor E2F (E2F4 or E2F5), a dimerization partner, and a multi-vulval class B (MuvB) repressor complex (Lin9, Lin37, Lin52, Lin54, and RBBP4), inhibits transcription of cell cycle regulators [67,70–72]. Phosphorylation of the Rb family protein p130 by cyclin-dependent protein kinases inhibits DREAM complex assembly, whereas inhibition of p130 phosphorylation by CDK blockade or phosphorylation of Lin52 at S28 by dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) facilitates DREAM complex assembly and promotes quiescence [71,73,74]. In addition, knockdown of the expression of Lin52 or DYRK1A [75], pharmacological inhibition of DYRK1A [76], or binding of proliferating cell nuclear antigen-associated factor to the core DREAM subunit RBBP4 [77] disrupted DREAM complex formation and caused cells to exit quiescence, indicating the important role of the DREAM complex in quiescence and cellular dormancy. Therefore, the DREAM complex could be a cellular target for regulating dormancy in cancer cells.
2. Regulation of cellular dormancy by interaction with the extracellular matrix
The ECM is a noncellular, physical, and structural component in tissues and organs that plays important roles in providing physical scaffolding and transducing cellular, biochemical, and biomechanical signaling pathways involved in the construction of cellular architectures, cellular polarity, cell proliferation, survival, motility, differentiation, and homeostasis [78–80]. More than 1,000 proteins that encode the ECM and ECM-associated proteins have been defined as “the matrisome,” and “the core matrisome,” which is composed of about 300 core ECM-associated proteins, contains repeats and/or various rearrangements of distinctive groups of domains, and constructs various types of ECM in a cell or tissue-specific manner [80,81]. Collagens, proteoglycans, and glycoproteins (such as laminins, fibronectin, and elastin) construct the core matrisome [80], and these ECM components transduce signaling via their receptors, including integrins, discoidin domain receptor tyrosine kinases (DDRs), syndecans, and dystroglycans [78–80].
ECM remodeling and concurrent changes in ECM-mediated signaling are believed to play an important role in the activation of dormant cancer cells, leading to recurrence and metastatic tumor formation [82]. For example, urokinase plasminogen activator receptor (uPAR), which has been reported to form a stable complex with integrins [83], interacts with integrin α5β1 and facilitates fibronectin fibril deposition, thereby leading to persistent ERK activation, p38 MAPK downregulation, and a consequent increase in cancer cell proliferation [84,85]. Downregulation of uPAR or blockade of uPAR and integrin α5β1 interaction causes dormancy in cancer cells [84]. In addition, fibronectin-mediated integrin β1 signaling causes myosin light chain kinase (MLCK)-mediated myosin light chain phosphorylation, leading to cytoskeleton rearrangement, stress fiber formation, and awakening of dormant cells [86]. Fibrotic circumstances mediated by type I collagen deposition also results in exit from dormancy and outgrowth of dormant cancer cells via the activation of integrin β1 signaling [87]. We have also demonstrated the involvement of upregulated type I collagen expression and integrin signaling in the awakening of slow-cycling/dormant cancer cells [88].
Studies have shown that ECM components contribute to cellular dormancy in cancer cells. For example, cells embedded in Matrigel exhibit dormant cell phenotypes [89]. In addition, dormant cancer cells induced by incubation with biomaterials under serum deprivation conditions produce type III collagen or fibronectin and create a type III collagen– or fibronectin-enriched matrix, resulting in the maintenance of cancer cell dormancy via activation of type III collagen–DDR1–STAT1 and ανβ3/α5β1 integrin-mediated signaling pathways [40,90]. Previous studies have shown that fibronectin-mediated integrin signaling activated focal adhesion kinase (FAK)–mitogen-activated protein kinase kinase (MEK)–ERK signaling is necessary for the survival of dormant cancer cells, which is in conflict with the role of ERK signaling in outgrowth but not the survival of dormant cancer cells, presumably due to the dynamics of ERK activity during the course of onset and maintenance of dormancy [40]. Moreover, an ECM glycoprotein, thrombospondin-1 [80], was elevated in dormant tumor cells [91] and promoted cellular dormancy in cancer cells by suppressing angiogenesis and activating transforming growth factor-β (TGF-β) [92]. The level of thrombospondin-1 showed an inverse correlation with tumor aggressiveness and a positive correlation with favorable clinical outcomes in cancer patients [93]. Another ECM glycoprotein, osteopontin, was also found to support dormancy in leukemia cells by providing an anchor for cell adhesion [94]; however, osteopontin was found to promote invasion and aggressiveness of tumor cells and may contribute to the awakening of dormant cancer cells via its ability to bind to integrin and activate integrin-mediated signaling [93,95,96], suggesting a different role of osteopontin in cellular dormancy in a context-dependent manner.
ECM stiffness, which is determined by the density of collagen and elastin and changes in their alignment via posttranslational modification [97,98], has also shown conflicting roles in cancer dormancy, depending on experimental models. A previous study demonstrated that cancer cells cultured on stiff supports displayed a proliferative phenotype by activating integrin β1-mediated ERK, Akt, and STAT3 signaling, whereas those cultured on soft supports exhibited dormancy phenotypes with elevated stemness-related marker expression, indicating the regulation of cell proliferation and dormancy by matrix stiffness [99]. Consistently, breast cancer cell spheroids grown on stiff hyaluronic acid (HA) hydrogels exhibited a proliferative phenotype, whereas those grown on soft HA hydrogels displayed a dormant phenotype [100]. In contrast to the proliferative phenotype, which is sensitive to anticancer therapy in breast cancer cells exposed to stiff microenvironments using synthetic substrata, cells exposed to soft microenvironments exhibited dormant phenotypes with increased autophagy and therapy resistance [101]. However, cells grown in a stiff fibrin matrix exhibit dormant phenotypes by the induction of p21 and p27 and downregulation of integrin β3 via the Cdc42-Tet2 pathway [102]. Considering the phenotypic and metabolic heterogeneity of dormant cancer cells [40,43,103], additional single cell–based in-depth investigations are necessary to determine the precise role of cell-ECM interactions in the maintenance or outgrowth of dormant cancer cells.
3. Regulation of cellular dormancy by environmental stresses and anticancer therapy
In addition to cell cycle regulatory and ECM-associated mechanisms, extracellular stress also mediates cellular dormancy. Dormant cancer cells, including DTCs, tend to encounter stressful microenvironments such as hypoxia and nutrient deprivation [103]. Cancer cells exposed to these environmental stressors display dormant phenotypes. For instance, hypoxia mediates cancer cell dormancy by the transcriptional upregulation of EMT and stemness phenotypes [104]. In addition, CSN8, a subunit of the constitutive photomorphogenesis 9 (COP9) signalosome, positively regulates hypoxia-induced EMT and cancer cell dormancy by stabilizing hypoxia-inducible factor 1-α (HIF-1α) and promoting HIF-1α signaling pathway activation [105]. Hypoxia-inducible gene domain family member 1A (HIGD1A), a HIF-1 target gene mediating cell survival, induced dormancy by repressing cellular respiration and reactive oxygen species (ROS) production [106]. Amino acid deprivation causes autophagy as a survival mechanism in dormant ovarian cancer cells through the upregulation of aplasia Ras homolog member I (ARHI or the Ras family member 3 [DIRAS3]) an autophagy inducer [107,108]. Glucose deprivation also induced autophagy, quiescence, and chemoresistance in glioblastoma cells [109]. In addition, serum deprivation caused cellular dormancy in cancer cells by fatty acid oxidation-mediated epigenetic Nanog expression and subsequent Nanog-mediated p21 and p27 induction [110]. In addition to these environmental stresses, cancer cells subjected to androgen-deprivation therapy [111], chemotherapy [42], or concurrent blockade of the epidermal growth factor receptor and MEK [112] were found to enter a dormant state. Activation of cellular stress responses, such as p53-mediated transcriptional response and integrated stress response, also mediated the spontaneous acquisition of slow-cycling phenotypes in cancer cells [113].
4. Regulation of cellular dormancy by p38 MAPK
Several mechanisms have been suggested to regulate cellular dormancy in response to environmental stress and anticancer therapies. Various environmental and cellular stresses cause cellular dormancy and activate several adaptive programs to endure cellular insults and maintain survival. Among the various signaling mediators, activation of p38 MAPK plays an important role in the induction of cellular dormancy and survival of dormant cancer cells [38,114]. p38 MAPK blocks cell cycle progression and induces quiescence by downregulating cyclins and upregulating CDK inhibitors (such as p21, p27, and p16) through the upregulation of nuclear receptor subfamily 2 group F member 1 (NR2F1), basic helix-loop-helix domain containing, class B, 3 (BHLHB3), and p53, and downregulation of c-Jun and forkhead box protein M1 (FoxM1) [38,115,116]. In addition, p38 MAPK exerts both proapoptotic effects by phosphorylation of the Bcl-2 family protein, Bim (EL), and activation of caspase-3, and prosurvival effects through increase in cyclooxygenase-2 (COX-2) and antiapoptotic inflammatory mediators [114,117–119]. Moreover, as a prosurvival mechanism of dormant cancer cells, p38 MAPK induces the nuclear translocation and activation of activating transcription factor 6 alpha (ATF6α), leading to the induction of Ras homolog enriched in brain (Rheb) and 78 kDa glucose-regulated protein (GRP78)/BiP/HSPA5 and, in turn, causes activation of mammalian target of rapamycin (mTOR) signaling and protection from Bax-mediated apoptosis [38,120–122]. Additionally, mitogen- and stress-activated protein kinase 1 (MSK1), a downstream target of p38 MAPK, controls markers of stemness and differentiation [123]. Thus, activation of p38 MAPK is essential for maintenance of cellular dormancy in various aspects.
5. Regulation of cellular dormancy by autophagy induction
Induction of autophagy, a self-clearance process that maintains cellular homeostasis by lysosomal degradation of malfunctioning organelles and unfolded or aggregated proteins [124,125], also plays an important role in the survival of dormant cancer cells. Among the three types of autophagy, including macroautophagy (generally known as autophagy), microautophagy, and chaperone-mediated autophagy [124, 125], only macroautophagy (autophagy) is known to contribute to cellular dormancy [125–127]. For example, autophagy plays a crucial role in the survival of gastrointestinal stromal tumor cells that undergo quiescence following treatment with imatinib mesylate [128] and the survival of disseminated breast cancer cells that enter a dormant state [129]. In addition, the tumor suppressor gene ARHI (also known as DIRAS3) was found to be critical for the survival of dormant ovarian cancer cells grown under in vivo conditions by inducing autophagy through inhibition of phosphoinositide 3-kinase/mTOR signaling, elevation of autophagy-related gene 4 (ATG4), along with the cleavage of light chain 3 (LC3) in autophagosomes [130]. DIRAS3/ARHI also maintains the survival of dormant ovarian cancer cells after treatment with conventional chemotherapeutic agents by facilitating the formation of the autophagosome initiation complex by binding to beclin 1 (BECN1) and inducing autophagy [131]. Moreover, autophagy has been found to be involved in the maintenance of diapause-like states in DTPs [41]. Despite the weak association of autophagy with slow-cycling/dormant cancer cells in our recent finding [42], these findings imply the essential role of autophagy in the control of cellular dormancy.
6. Regulation of cellular dormancy by growth factors
Several growth factors, derived autonomously or from stromal cells in the surrounding microenvironment, are also involved in the induction and maintenance of cancer cell dormancy. The TGF-β superfamily consists of more than 30 members of growth factors that play a pivotal role in cell proliferation, growth, survival, and differentiation, playing both tumor-suppressive and tumor-promoting roles in cancer [132]. Recent studies have demonstrated the role of TGF-β2 and bone morphogenic protein-7 (BMP-7) in cancer cell dormancy. TGF-β2 derived from the microenvironment in bone marrow (BM) caused dormancy of DTCs through TGFβR3-mediated p38 MAPK activation, p27 induction, and CDK4 downregulation [133]. Growth arrest specific 6 (Gas6)/Axl signaling and the consequent induction of TGF-β2/TGFβR also induced dormancy in disseminated prostate cancer cells [134]. In addition, BMP-7 derived from stromal cells in the BM microenvironment caused dormancy of prostate cancer cells through the activation of BMP-7/BMPR2–mediated upregulation of p38 MAPK and N-myc downstream-regulated gene 1 (NDRG1) [135]. Moreover, insulin-like growth factors (IGFs), which are known to play an important role in the growth, proliferation, survival, differentiation, and metastasis of cancer cells [136], have been found to be associated with cancer cell dormancy. After ablation of oncogenic drivers, dormant residual pancreatic cancer cells (mutant Kirsten Rat Sarcoma Viral Oncogene Homolog [KRAS] [KRASG12D/+] and c-MYC) maintain their survival by the autocrine activation of IGF1/IGF-1R and downstream Akt signaling as a compensatory mechanism [137]. IGF2 also caused dormancy and chemoresistance in osteosarcoma cells by downregulating the conventional IGF-1R/Akt signaling and enhancing autophagy and glutamine utilization [138]. Given the impact of IGFs on the promotion of cancer cell proliferation [136] and the context-dependent modulation of cell proliferation by IGF1 [139], additional studies are necessary to elucidate how IGFs determine cell fate (dormancy or proliferation) under certain circumstances.
7. Epigenetic mechanisms
Dormant cancer cells are able to intersperse dormancy and proliferation through epigenetic reprogramming mechanisms, including DNA methylation and histone modifications [54,55]. The expression of orphan nuclear receptor NR2F1 is suppressed in various cancers through promoter hypermethylation but becomes highly expressed during dormancy [56]. NR2F1 induces global chromatin repression by activating NANOG, leading to dormancy of DTCs in the BM [116]. Additionally, transcription factor SOX9, retinoic acid receptor β, and CDK inhibitors are known to mediate NR2F1-induced quiescence [116]. Concerning deregulation of microRNAs, a known epigenetic modulator [140], a consensus signature of human tumor dormancy-associated miRNAs (DmiRs) has been identified in human dormant breast carcinoma, glioblastoma, osteosarcoma, and liposarcoma tumors [141]. Moreover, a stable microRNA (miRNA, miR) switch has been shown to regulate dormant to proliferating phenotype transition [141]. For example, it has been shown that upregulation of miR-101 concurrently activates a number of molecules associated with dormant CSC phenotype, such as enhancer of zeste homolog 2 (EZH2)- and TP53- related proteins [142]. In addition, overexpression of miR-90 causes the formation of dormant microtumors in glioblastoma and osteosarcoma cells and inhibits tumor progression by modulating transcription factors, tumor suppressor genes, and interferon response pathways [143]. Hence, microRNAs as well as DNA or histone modifications are also implicated in the regulation of cellular dormancy.
8. Regulation of cellular dormancy by control of endoplasmic reticulum stress-mediated unfolded protein response
Environmental stresses, including hypoxia and glucose deprivation, and deregulated ECM-mediated signaling are known to disrupt homeostasis in the endoplasmic reticulum (ER) [45,144], resulting in the induction of ER stress and subsequent activation of the unfolded protein response (UPR), which determines cell fate depending on the duration and/or magnitude of stress [145]. Among the three branches of UPR, protein kinase R-like endoplasmic reticulum kinase (PERK), ATF6α, and inositol requiring enzyme 1 alpha (IRE1α), ATF6α and IRE1α branches mediate the clearance of misfolded proteins from the ER by inducing transcription of genes regulating protein folding or degradation (ER-associated protein degradation, ERAD) in the ER, whereas the PERK branch causes global attenuation of protein translation by phosphorylation of eukaryotic initiation factor 2 (eIF2α) to reduce the load of misfolded proteins and facilitate the repair of ER homeostasis [146,147]. The processing (cleavage) of ATF6 by site-1 and site-2 proteases (S1P and S2P) in the Golgi apparatus and the splicing of X-box binding protein 1 (XBP1) through IRE1α are required for transcriptional activity and full activation of the UPR [148]; IRE1α also mediates regulated IRE1-dependent decay of mRNA (RIDD), resulting in both survival and death of the cells by preserving ER homeostasis and causing the decay of pre-microRNAs, respectively [149]. In addition, the PERK-eIF2α pathway eventually induces ATF4, thereby regulating cell survival and death via growth arrest and DNA damage-inducible protein (GADD) 34-mediated eIF2α dephosphorylation and GADD153/CHOP-mediated apoptosis [146,147]. However, protein overload through ATF4 overexpression also caused ROS production and apoptotic cell death [150]. Dormant cancer cells regulate UPR for survival. For example, the UPR is activated in DTCs and mediates their survival against hypoxic and glucose deprivation [151]. In addition, disseminated pancreatic cancer cells exhibit elevated PERK pathway activation with diminished activation of the IRE1α pathway, resulting in the acquisition of a quiescent phenotype and escape from CD8+ T cell–mediated antitumor immunity via lack of major histocompatibility complex class I (MHCI) expression [152]. Activation of p38 MAPK protects dormant cancer cells from chemotherapy-induced ER stress by increasing GRP78/BiP expression and PERK activation [153].
9. Regulation of cellular dormancy by control of ER stress–mediated protein translation
Translational control during ER stress is crucial for maintaining cellular homeostasis and promoting cell survival [154], and, as described above, dormant cancer cells utilize UPR for survival. However, the underlying mechanisms remain to be determined. We have found altered regulation of the UPR in slow-cycling/dormant cancer cells that were identified according to CSFE dye retention or enriched by chronic treatment with chemotherapeutic agents [42]. These SCC cells exhibited typical dormancy-like phenotypes, such as an elevated p38 MAPK/ERK ratio (p38High/ERKLow), decreased expression of cyclins and CDKs, and increased expression of CDK inhibitors (p21 and p27) without senescence, stemness, and EMT-like phenotypes [42]. In addition, these SCC cells displayed upregulation of ATF6 and IRE1 target genes (such as chaperones and ERAD-associated genes) and downregulation of ATF4 protein and its target genes, such as GADD34 and CHOP, with elevated PERK and eIF2α phosphorylation [42]. Mechanistically, the regulator of G protein signaling 2 (RGS2), a GTPase-activating protein (GAP) [155], was elevated in these SCCs, causing ubiquitin-mediated degradation of ATF4 by direct binding and sustained translational arrest [42]. These findings imply that the regulation of UPR machinery for ER homeostasis is involved in the survival of dormant cancer cells upon sustained ER stress, such as chemotherapy. In addition, along with the GAP-independent role of RGS2, which includes translational control [156] and a component of cellular stress response [157–159], these findings suggest a novel function of RGS2 in the survival of dormant cancer cells by maintaining protein homeostasis against sustained ER stress caused by chemotherapy or hostile microenvironments (Fig. 3).
Awakening of Dormant Cancer Cells
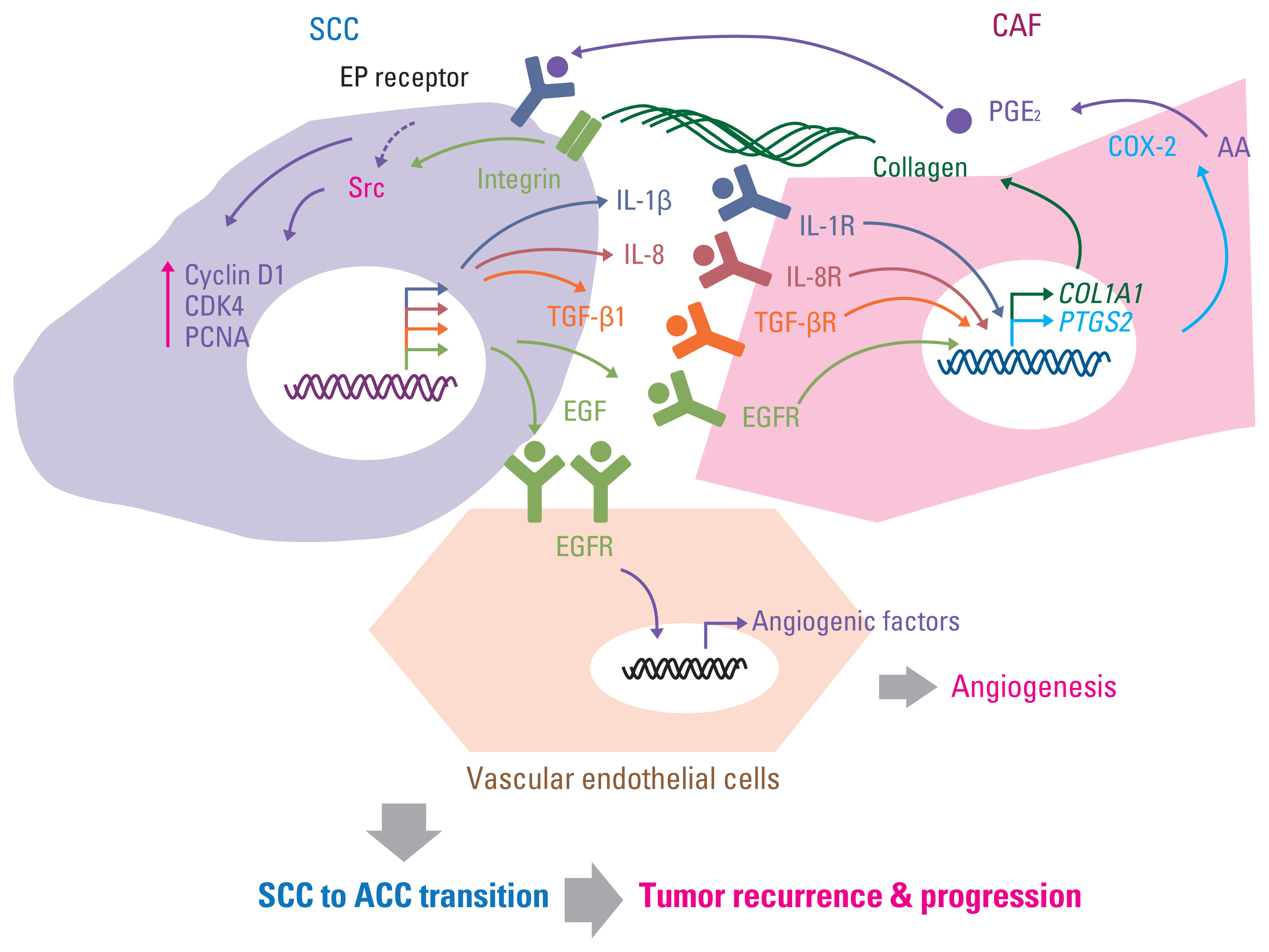
As discussed in previously published literature in more detail, the breakdown of the aforementioned maintenance mechanisms of cellular dormancy under the coordinated influence of several factors, including ECM composition, direct or soluble factor-mediated indirect interaction with surrounding stromal cells in the microenvironment, nutritional availability, chronic inflammation, and other host factors, mediates the awakening of dormant cells and the formation of recurrent tumors [13,24,43,160]. In addition to the autonomous changes of autocrine soluble factors [161], the interaction with stromal cells, such as myeloid cells (macrophages, myeloid-derived suppressor cells [MDSCs], and neutrophils), pericytes, fibroblasts, and vascular endothelial cells via growth factors, cytokines, and chemokines, plays a crucial role in recurrent tumor formation [13,24,160]. For example, tissue-resident macrophages in the mammary gland are a major source of tumor-associated macrophages and mediate local recurrence and distal metastasis of triple-negative breast cancer [162]. The increase in C-X-C motif chemokine ligand 10 (CXCL10) and induced mobilization of toll-like receptor 4 (TLR4) receptor-expressing monocytic MDSC through the upregulation of matrix metalloproteinase 14 (MMP14) has also been suggested as a mechanism of hepatocellular carcinoma recurrence after liver transplantation [163]. In addition, sustained inflammation caused by bacterial infection or tobacco smoking induces the formation of neutrophil extracellular traps and extracellular DNA scaffolds bound to ECM, such as laminin, and contains cytotoxic proteins and proteases (such as neutrophil elastase and matrix metalloproteinase 9), consequently evoking the proliferation of dormant cancer cells in the lungs by the activation of integrin-mediated FAK/ERK/MLCK/Yes-associated protein 1 (YAP) signaling through a series of sequential protease-mediated laminin remodeling [164]. Activated hepatic stellate cells, the liver-specific pericytes, were found to awaken dormant breast cancer cells in the liver by releasing CXCL12 and causing CXCR4-mediated quiescence in NK cells and CXCR4-induced outgrowth of dormant cancer cells [19]. Moreover, fibroblast proliferation and microvessel formation were found to exist prior to recurrent tumor formation after radiation [165], and several studies including ours demonstrated the outgrowth of SCCs/dormant cancer cells by increasing the recruitment of fibroblasts and vascular endothelial cells under cytokine- and growth factor–mediated proinflammatory and proliferation-promoting circumstances [88,166,167] (Fig. 4). In summary, the interaction with the surrounding microenvironment can be a cue for awakening dormant cancer cells and outgrowth of recurrent tumors, and strategies targeting these interactions can prevent recurrent tumor formation.
Targeting Dormant Cancer Cells
The development of therapeutic approaches targeting dormant cancer cells is significant. Based on recent findings on the regulatory mechanisms of cellular dormancy and reactivation, several strategies for targeting cancer cell dormancy and blocking recurrent tumor formation have been suggested [11–13,24]. In a recent paper, Recasens and Munoz suggested three potential strategies, termed as “Sleeping strategy,” “Awakening strategy,” and “Killing strategy” to maintain, awaken, or eradicate of dormant cancer cells [12]. The sleeping strategy prevents dormant cancer cells from entering the proliferative status [12] by suppressing integrin-mediated proliferative signaling pathways (for example uPAR, β1 integrin, MLCK, Src, and ERK) [84,86,87], epigenetically inducing dormancy-associated factors (such as induction of NR2F1 by treatment with 5-Aza-C, a DNA methyltransferase inhibitor, either alone or in combination with all-trans-retinoic acid) [116], and treating dormancy-inducing soluble factors, such as TGF-β2 and BMP-7 [133,135]. The awakening strategy makes dormant cells vulnerable to anticancer therapy that targets proliferative cells by forcing them to enter the cell cycle [12]. In our previous study, genomic ablation or RGS2 expression or an increase in protein translation by treatment with a clinically available phosphodiesterase 5 (PDE5) inhibitor (sildenafil, for example) induced the proliferation of SCC/dormant cells but enhanced the antitumor efficacy when combined with chemotherapeutic agents [42]. Despite the possibility of aggressive recurrent tumor formation at the primary and distal sites [12], this strategy may be effective in combination with appropriate anticancer therapeutics. Finally, the killing strategy eliminates dormant cancer cells [12]. For example, treatment with IFN-γ combined with an inhibitor of IDO1 or AhR inhibits dormant cancer cells [102]. In addition, in a pancreatic cancer model, inhibition of IGF-1R reduced MRD [137]. Targeting Unc-51 like autophagy activating kinase 1 (ULK1) combined with CPT-11 can also diminish the regrowth of dormant cancer cells [41]. In the case of dormant cancer cells with senescence-like phenotypes, using senolytic drugs that target senescent cells could be a strategy to kill dormant cancer cells [24]. In our previous study, treatment with chemotherapy in combination with clinically available Src or COX-2 inhibitors inhibited the growth of SCC/dormant cancer cells [88]. Examples of targeting dormant cancer cells are listed in Table 2. Because potential targeting strategies for dormant cancer cells have both advantages and disadvantages [12], a combinatorial approach utilizing one of these strategies would be appropriate to achieve complete elimination of dormant cancer cells. In addition, the development of new drugs that modulate a new dormancy-associated cellular target, or those with better efficacy and reduced toxicity, will be promising. Moreover, repurposing existing clinically available drugs, as demonstrated in our recent publications [42,88], would be beneficial for developing clinically relevant therapeutic strategies.
Conclusions and Perspective
Tumor dormancy is a critical step in cancer development and drug resistance [11,13,24,160]. Mechanistic insights into cellular and tumor dormancy are essential to understand how tumor cells become dormant or awaken, to prevent tumor relapse and progression, and to maximize therapeutic benefits. Despite its importance, understanding the biology of tumor dormancy has been limited because of the lack of appropriate methodologies to model dormancy in experimental models or to detect dormant cancer cells in clinical samples. However, in recent decades, several experimental approaches to mimic dormancy have been developed, including the use of genetically engineered mouse models [37] and technologies to detect dormant cancer cells using tissue or liquid biopsies [168,169]. Given the heterogeneity of dormant cancer cells and their cellular dynamics [40], such as cellular and phenotypic heterogeneity and plasticity in EMT [170,171], it is likely that a plethora of intracellular and extracellular factors rewire proliferative and metabolic status in dormant cancer cells through highly dynamic processes in cooperation with various components in the microenvironment at primary and metastatic sites. Therefore, the answers are likely to be complicated. Further studies are required to identify dormancy-associated cellular markers using in-depth investigation of dormant cancer cells at the single-cell level. Such advances will aid in a better understanding of the mechanisms involved in the course of entry and exit from cellular dormancy and prevent the development of recurrent tumor formation through the diagnosis of dormant cancer cells at an early stage. These endeavors would help protect cancer survivors from fatal clinical outcomes and lower the health and socioeconomic burden of cancer.