Introduction
Erdheim-Chester disease (ECD), first described in 1930, is an extraordinary form of non-Langerhans cell histiocytosis (LCH) [1]. The reported number of confirmed patients is about 500, and reports have recently increased. The clinical manifestation of ECD varied from indolent disease to a devastating course associated with significant organ damage to the heart, lung, bone, and central nervous system [2]. Nevertheless, ECD is often not suspected because most patients usually present with nonspecific symptoms such as fever, weakness, weight loss, and night sweating. Moreover, there are no widely accepted diagnostic guidelines for ECD because of limited experience with this rare disease.
LCH and ECD share a few pathological and clinical manifestations. Pathologically, LCH is characterized mainly by proliferation of CD68 positive, CD1a positive, and S100 positive dendritic cells. Clinical features of LCH include lytic axial bones, peribronchial lung involvement, and scaly erythematous patch infiltration in the skin. However, ECD shows proliferation of non-dendritic phagocytes, which are commonly CD68 positive, CD1a negative, and S100 negative [3]. Clinically, ECD demonstrates appendicular bony sclerosis and myocardial infiltration. BRAF V600E mutation, which is associated with the production of pro-inflammatory cytokines and systemic inflammation, was reported in LCH at a frequency less than 50% and in ECD at that greater than 50% [4,5]. It is not easy to discriminate between the two diseases based on clinical features, so pathologic confirmation is mandatory.
The standard treatment for ECD is not established because of the lack of large-scale clinical trials. Some investigators have suggested that mitigating cytokine disturbances and balancing these cytokines could be an effective measure [6]. In addition, a few researchers have suggested that disease evolution seemed to be associated with T helper-1-induced pro-inflammatory chemokines, such as interleukin-6 and interferon-α (IFN-α) [7–9]. Therefore, they recommended that IFN-α, pegylated-interferon alfa 2a (PEG-IFN-α 2a), and ropegylated-interferon α-2b (ROPEG-IFN-α 2b) could be a therapeutic option for advanced ECD [10,11]. ROPEG-IFN-α 2b has recently become available due to discontinued PEG-IFN-α 2a production a few years ago. Unfortunately, there are few data on ROPEG-IFN-α 2b in patients with ECD.
Herein, we share two cases treated with different IFN-α, PEG-IFN-α 2a and ROPEG-IFN-α 2b. Based on the case reviews, we discussed the current role of ROPEG-IFN-α 2b in the absence of PEG-IFN-α 2a for ECD patients. This study was approved by the Institutional Review Board (IRB) of Samsung Medical Center and was performed under the relevant guidelines and regulations, including the Declaration of Helsinki. All patients provided informed consent under the IRB regulations (NCT #03117036).
Case Report
1. Case 1
A 70-year-old woman had a 2-year history of type 2 diabetes mellitus, hypertension, and coronary artery disease. Moreover, she had undergone coronary artery bypass surgery 1 year prior and received implantable cardioverter-defibrillator insertion due to ventricular fibrillation 4 months prior.
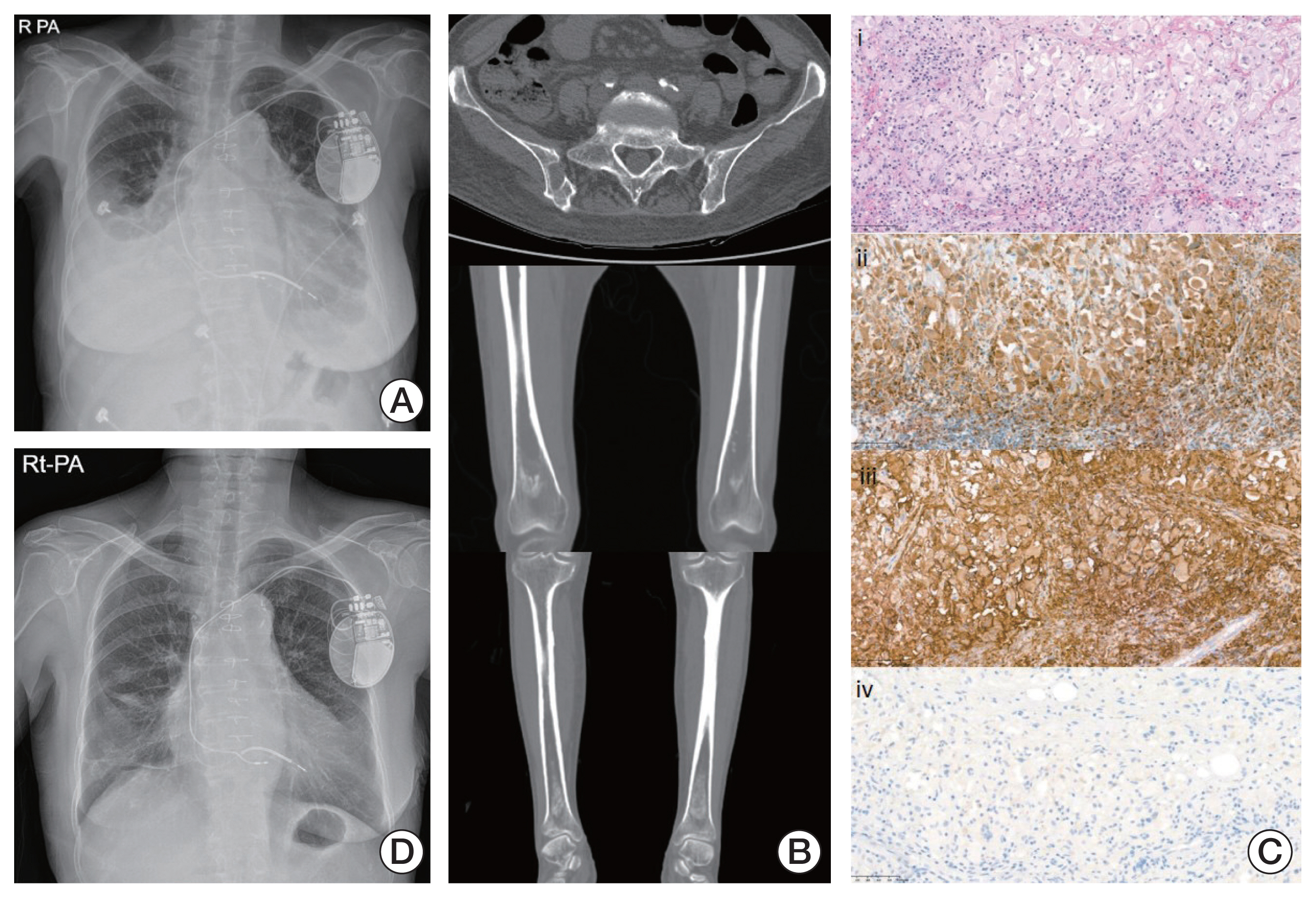
She was admitted to the emergency room complaining of persistent generalized edema with pretibial pitting edema, dyspnea, intermittent chest pain, and general weakness and was estimated to have an Eastern Cooperative Oncology Group performance score of 4. Chest X-ray showed severe cardiomegaly with bilateral pleural effusion and pulmonary edema (Fig. 1A). Based on chest computed tomography scan (CT) and transthoracic echocardiography, diffuse right atrial wall thickening was discovered. Moreover, skeletal CT showed osteolytic and osteosclerotic lesions in multiple weight-bearing bones (Fig. 1B). Under the suspicion of cardiac malignancy, open cardiac biopsy in the right atrial mass was performed. The final pathology results showed foamy histiocytes stained strongly with CD68, CD163, and S-100 but not CD1a, and a BRAF V600E mutation suggested a diagnosis of ECD with BRAF V600E mutation (Fig. 1C).
The patient was administered PEG-IFN-α 2a 180 μg once weekly for 6 months and then every 2 weeks for the next 4 months. The subjective symptoms, especially edema, dyspnea, chest pain, and objective findings such as pleural effusion and cardiomegaly dramatically improved after 3 months of PEG-IFN-α 2a injection (Fig. 1D). The most common adverse events of PEG-IFN-α 2a administration were fatigue, general weakness, and decreased appetite, which were mild and manageable. After the induction period for rapid disease control, the clinicians suggested that she receive PEG-IFN-α 2a every 4 weeks for the next 10 months as consolidation treatment. The disease was well controlled at 20 months, and the administration schedule was changed to once every 2 months as maintenance therapy.
2. Case 2
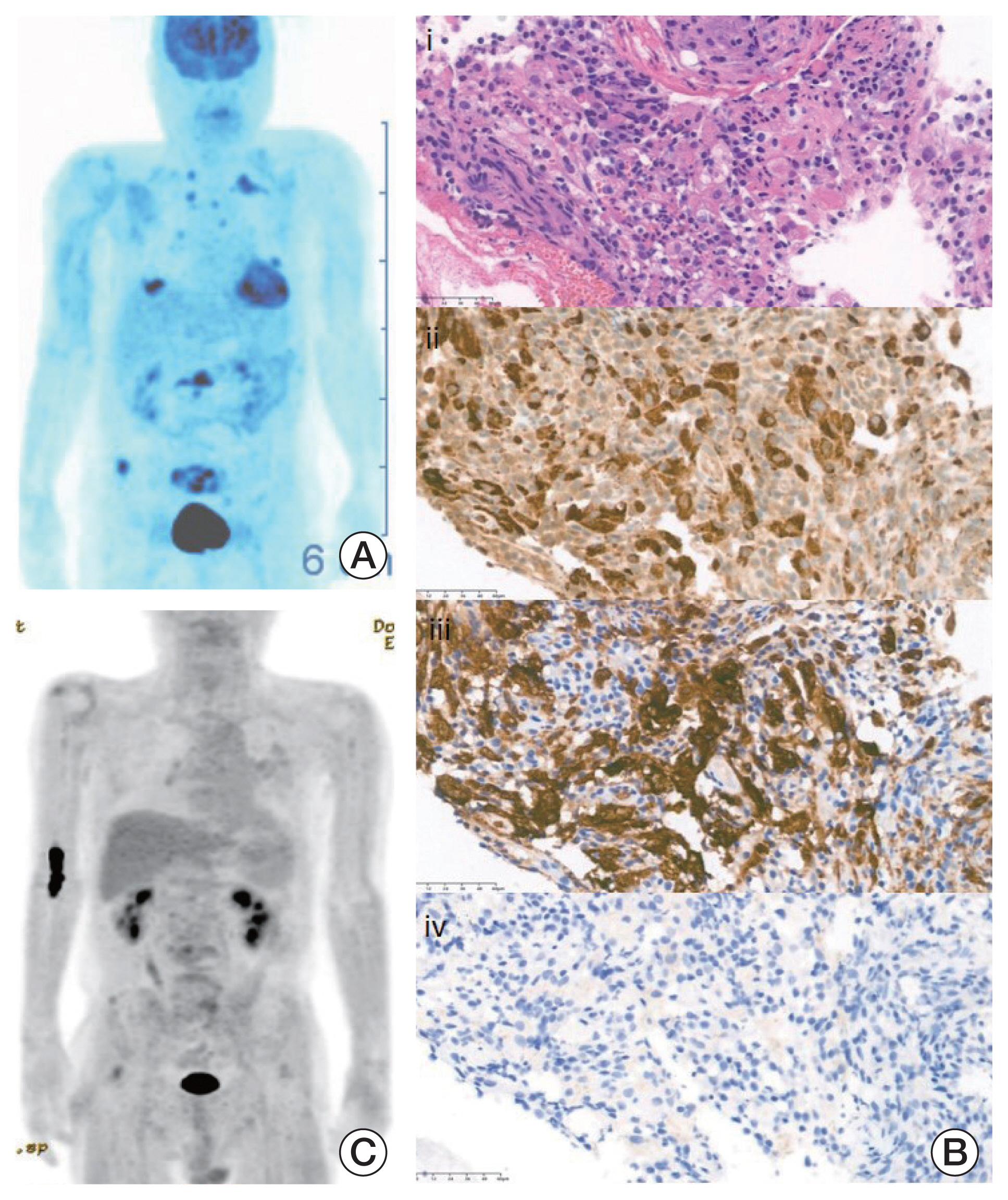
A 65-year-old man who suffered from a 2-month history of back pain was referred to the hematology-oncology department for suspected hemophagocytic lymphohistiocytosis (HLH). He presented with fever, pancytopenia (absolute neutrophil count 18,290/mm3, hemoglobin 8.3 g/dL, platelet 69,000/mm3), hyperferritinemia (1,009 ng/mL), organomegaly, and hyper-triglyceridemia (226 mg/dL), and prominent hemophagocytosis was found in bone marrow aspiration. In spine magnetic resonance imaging and positron emission tomography computed tomography (PET-CT) scan, multiple osteolytic lesions in the lumbar spine were detected (Fig. 2A). The pathologic review of the bone biopsy revealed CD68 and CD163 positivity but CD1a negativity in numerous proliferating histiocytes. Considering these pathologic findings, ECD without BRAF V600E mutation was diagnosed in next-generation sequencing (Fig. 2B).
Due to discontinuation of PEG-IFN-α 2a, the patient started ROPEG-IFN-α 2b 500 μg/mL subcutaneous injections once a month. ROPEG-IFN-α 2b is a drug that can be initially administered every 2 weeks and then increased every 4 weeks later. However, the patient was administered ROPEG-IFN-α 2b once a month from the beginning in consideration of his economic situation. Clinical features of HLH resolved at 3 months, and multiple bone pain significantly improved after four injections. The patient received ROPEG-IFN-α 2b for 8 more months as an induction, maintaining an excellent clinical response without specific toxicity. In addition, multiple high standardized uptake value uptake lesions observed in baseline PET-CT nearly resolved in follow-up imaging (Fig. 2C). Thus, the dosing interval of ROPEG-IFN-α 2b was increased to every 2 months as a consolidation treatment.
Discussion
ECD, a rare non-LCH, is a multi-systemic disease with unclear pathogenesis. There are limited data in the management of this disease based on a handful of retrospective or pilot studies. Currently, IFN-α/PEG-IFN-α 2a has been used as the front-line treatment option for patients with ECD, with reported prolonged survival outcomes [12,13]. In the present cases, we used PEG-IFN-α 2a and ROPEG-IFN-α 2b for two severe ECD patients as both have been effective irrespective of BRAF V600E mutation status.
There is no established optimal dose or schedule in PEG-IFN-α 2a and ROPEG-IFN-α 2b. According to the drug excretion half-life, we started treatment with once weekly PEG-IFN-α 2a and once monthly ROPEG-IFN-α 2b for rapid disease control. At the time of response, we reduced dosing schedules to every 2 weeks or once a month for PEG-IFN-α 2a and every 2 months for ROPEG-IFN-α 2b for consolidation. Interestingly, both patients achieved a sustained response and showed no severe adverse events during the treatment period. Although we cannot recommend an optimal dose or schedule of IFN for ECD patients, it is reasonable to start IFN-α intensively for rapid symptom control at the early phase of the disease and decrease the dose intensity after achieving response.
There are no standard guidelines for response evaluation for ECD. Thus, it is challenging to modulate an injection schedule and cessation of treatment based on the response. Munoz et al. [2] reported that the median treatment duration of IFN was 23 months, and a few patients maintained IFN treatment for a long time until disease progression. In our report, ECD patients showed symptom improvement at 3 to 4 months after starting treatment. We recommend monitoring symptom improvement monthly and conducting imaging studies every 3 months.
The monopeglyated IFN-α ROPEG-IFN-α 2b has been developed for patients with myeloproliferative neoplasms. This drug has an extended elimination half-life, enabling less frequent injections, and has fewer toxicities [11,13]. Its efficacy and toxicities have not yet been reported in ECD patients. Our case 2 received ROPEG-IFN-α 2b and achieved complete radiologic response and significant symptom improvement after 8 months of treatment. ROPEG-IFN-α 2b seems to be an excellent alternative agent for newly diagnosed ECD patients as a substitute for PEG-IFN-α 2a. However, further research is needed to establish the optimal dose and schedule for ROPEG-IFN-α 2b.
Several chemotherapeutic modalities have been explored in the treatment of ECD. A BRAF inhibitor, vemurafenib, is another treatment option for ECD patients with BRAF V600E mutation as a salvage therapy. Although vemurafenib demonstrated an impressive response, BRAF V600E mutation is found in only about 50% of patients, and the drug is not effective in BRAF wild type. The purine analog cladribine has been used in patients as frontline or salvage therapy. Cladribine has shown overall response rates of about 50% and a response duration of 9 months [5,14]. Although cladribine was quite active in BRAF V600E mutation-negative patients, the duration of response was relatively short. Therefore, more promising new agents for ECD are needed [15].
In conclusion, PEG-IFN-α 2a and ROPEG-IFN-α 2b were very effective and tolerable for management of patients with ECD.