Introduction
Hepatocellular carcinoma (HCC) is the fifth most commonly diagnosed cancer and the second most frequent cause of cancer death in men worldwide [1]. The treatment algorithms in the American Association for the Study of Liver Disease (AASLD) and the European Association for the Study of the Liver (EASL) and the European Organization for Research and Treatment of Cancer (EORTC) guidelines recommend resection and ablation or transplantation for early-stage HCC, transcatheter arterial chemoembolization for intermediate-stage HCC and sorafenib (SR) for advanced-stage HCC [2,3]. Unfortunately, cancer cells often develop chemoresistance, which presents a major obstacle to the long‐term efficacy of chemotherapeutic treatments. There is increasing evidence that resistance to chemotherapy is, at least in part, caused by inherent resistance of a subpopulation of cancer cells, which have been labeled as cancer stem cells (CSCs) [4]. Liver CSCs are considered to be responsible for tumor growth, metastasis and recurrence of HCC as well as for the failure of chemotherapy and radiotherapy [5]. The traditional strategies leave behind residual CSCs which possess the abilities of self-renewal, multi-directional differentiation, and indefinite proliferation, as well as high tumorigenic ability [4], leading to cancer recurrence and metastasis. Therefore, the cancer stem cell theory offers novel insight into tumor diagnosis, treatment, and prevention.
Several markers have been proposed in the literature to identify CSCs in HCCs using cell surface antigens [6,7]. Recently, Haraguchi et al. [8] identified CD13, a membrane-bound zinc-dependent type II exopeptidase, as a candidate liver cancer stem cell marker by a surface marker screen based on microarray analysis. CD13+ CSCs were enriched in side population, which is known as the multidrug-resistant cell fraction with ATP-binding cassette transporter expression, and also localized predominantly in G1/G0 phase [8,9]. Thus, CD13+ cells represent the dormant or slow-growing population that is believed to account for the chemoresistant capacity in HCC. In this study, we observed selective enrichment of CD13+ cells following chemotherapies in HCC, suggesting that CD13+ CSCs are major reservoir for eventual recurrence for HCCs. Suppressing residual CD13+ CSCs after initial tumor debulking may sustain remissions and extend the progression-free survival of patients receiving CSC suppressive therapy. However, this task is complicated by the fact that CD13 is widely distributed in many tissues of mammals, such as the intestine, kidney, and liver as well as the central nervous system [10]. Until these challenges are overcome, CSC-targeting therapies will not reach their full potential.
Emerging evidence suggests metabolic reprogramming as a key player in CSC biology [11]. The action of metabolic pathways in CSC maintenance might not be merely the consequences of genomic alterations. Indeed, the metabotypic phenotypes may play a causative role in maintaining the stem traits. Therefore, targeting the metabolic events represents a potential strategy to halt tumor recurrence and refractoriness to treatment [12,13]. As opposed to differentiated bulk tumor cells which can be highly glycolytic or oxidative phosphorylation dependent, it is difficult to univocally identify the metabolic phenotypes that maintain CSC stemness. Due to the different microenvironmental conditions they survive in, CSCs are most likely supposed to get energy from different sources, according to substrate availability [11]. Based on the facts above, we further conducted a series of experiments to examine the metabolic phenotypes that render CD13+ CSCs multidrug-resistant in HCCs.
Materials and Methods
1. HCC cell lines
Huh7 and HepG2 cells were obtained from American Type Culture Collection and cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen).
2. Flow cytometry and cell sorting
Cells were incubated in phosphate-buffered saline containing 2% fetal bovine serum with phycoerythrin-conjugated anti-human CD13 antibody (Becton Dickinson, San Jose, CA). Isotype-matched mouse immunoglobulins served as controls. For flow cytometry, samples were analyzed using a FACSCalibur flow cytometer and CellQuest software (BD Biosciences, San Jose, CA). For cell sorting by flow cytometry, samples were analyzed and sorted on a BD FACSVantage SE (BD Biosciences). For the positive and negative population, only the top 25% most brightly stained cells or the bottom 20% most dimly stained cells were selected, respectively. Aliquots of CD13+ and CD13− sorted cells were evaluated for purity with a FACSCalibur machine and CellQuest software (BD Biosciences), using PE-conjugated anti-human CD13 antibody.
3. In vitro isotope labeling and kinetic profiling
To trace glucose, glutamine or tyrosine metabolism, CD13+ cells were grown as tumorspheres in growth media (Dulbecco's modified Eagle's medium/F12 nutrient mixture Ham [DMEM/F12]) on 10 cm dishes and then transferred into glucose-, glutamine- or Tyr-free DMEM/F12 medium supplemented with 13C-glucose, 13C-glutamine, and 13C-tyrosine (Cambridge Isotope Labs, Tewksbury, MA) to 10 mM (for glucose), 2 mM (for glutamine), or 0.1 mM (for Tyr) overnight (for steady-state labeling) or for the indicated time points in the flux analyses. Additionally, fresh media containing 13C-glucose, 13C-glutamine, and 13C-tyrosine was exchanged 2 hours prior to metabolite extraction for steady-state analyses.
4. Statistical analysis
All statistical analyses were carried out with SPSS ver. 22 (IBM Corp., Armonk, NY). ANOVA, Student’s t test, and Dunnett’s multiple comparison tests were used to compare mean values. Data are presented as mean±standard deviation. A p-value of < 0.05 was defined as statistically significant. Other methods are shown in Supplementary Methods.
5. Ethical statement
Human HCC specimens were obtained with written informed consent from all subjects before surgery and in accordance with the ethical standards of the Institutional Review Board of HuaShan Hospital (No. 20170036). The collection and processing of all samples were carried out in accordance with the Declaration of Helsinki. All mouse experiments were conducted in accordance with standard operating procedures approved by the Institutional Animal Care and Use Committee for Shanghai Public Health Clinical Center (protocol No. 2018X112) and adhered to the Animals in Research: Reporting In Vivo Experiments (ARRIVE) Guidelines.
Results
1. Selective enrichment of quiescent CD13+ CSCs following chemotherapy in HCCs
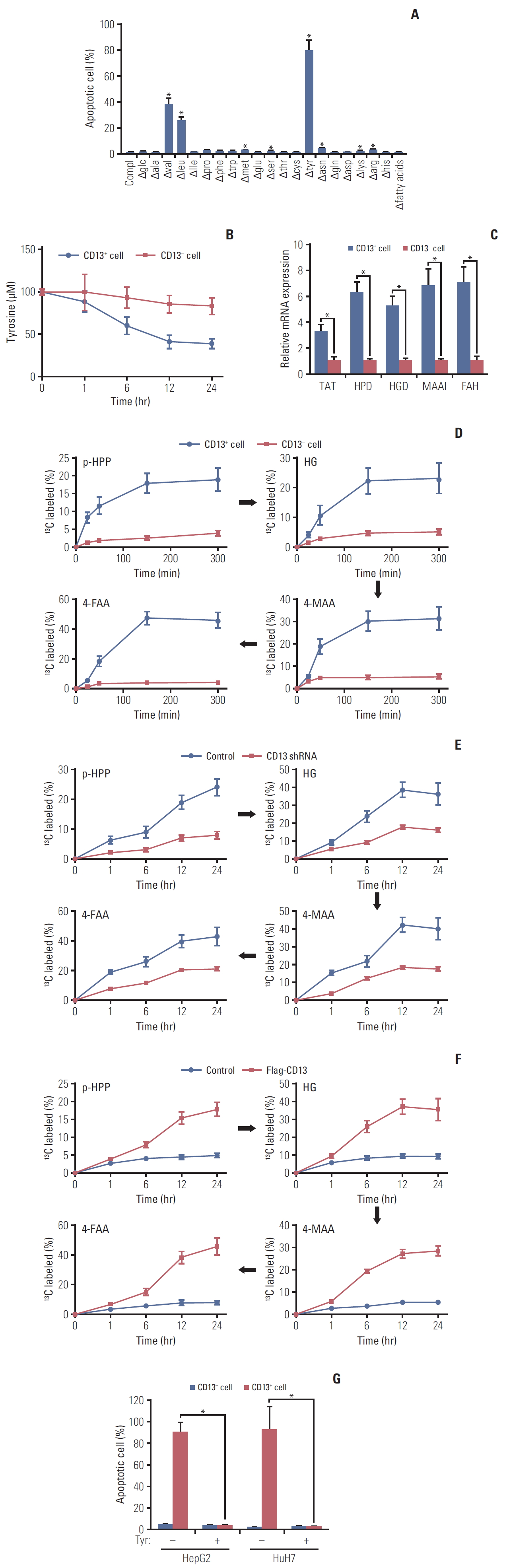
Analysis of HepG2 cells by flow cytometry showed that CD13- cells were a majority (82.3%±6.9%) of the total population before treatment. However, the viable CD13- population was reduced to 15.3%±3.1% after exposure to 5-fluorouracil (5-FU; 5 μM) for 72 hours, while the viable CD13+ population increased from 8.4%±2.3% to 72.1%±5.2% (Fig. 1A left). Similar results were observed in HepG2 cells after exposure to SR (4 μM) (Fig. 1A left). Immunoblotting further confirmed a significant enrichment for endogenous CD13 protein (Fig. 1B). Similarly, neither 5-FU nor SR failed to eradicate all HCC cells and enriched for CD13+ cells in HuH7 (Fig. 1A right and B). The quiescent state of CSCs is believed to protect them from therapeutic agents [14]. As compared with CD13- cells, the matched CD13+ subpopulation expressed lower levels of markers for cell proliferation, including Ki67, proliferating cell nuclear antigen, and MCM2 (Fig. 1C). Analysis of HepG2 and HuH7 cells using Hoechst plus pyroninY staining showed that > 60% of CD13+ fraction were in the G0 phase. In contrast, most CD13- fractions were actively cell cycling (< 40% in the G0 phase) (Fig. 1D). In addition, we examined changes in CD13 staining in nine HCC patients after receiving hepatic arterial infusion chemotherapy, using low-dose concomitant radiochemotherapy with cisplatin (CDDP) and 5-FU with/without leucovorin. All patients initially achieved a partial regression to drug treatment. As compared to the pretreatment values, the score of CD13 staining was significantly increased in seven out of nine residual tumors (Fig. 1E and F).
Three-dimensional (3D) organoids serve as an excellent tool to recapitulate tumorigenic aspects and drug responses of advanced human carcinomas [15]. In the current study, we generated 3D organoids from therapy-naive patients with HCC. Briefly, single-cell suspensions from tumor biopsies were suspended in Matrigel with fully supplemented aDMEM/F12+. Spherical structures, which were defined as 3D organoids, were detected by day 7 and continued to grow to ~200 μm in diameter by day 14 (Fig. 1G). We successfully generated 3D organoids from three of five tumors (60%), two (T3 and T4) of which could be passaged at least once to exhibit a continuous growth (Fig. 1H). Quantitative analysis demonstrated that the organoids harbored similar percentages of CD13+ cells when compared with the corresponding primary tumors (Fig. 1I). Moreover, the proportion of CD13+ cells remained relatively constant in organoids during serial passages (Fig. 1J), suggesting that organoids contain steady ratios of CSC populations in long-term cultures. Next, we evaluated the efficiency of organoid formation after treatment of chemotherapeutic agents. Upon administration of 5-FU or sorafenib, the efficiency of organoid formation decreased; this impairment remained unchanged along with serial passages of organoid culture (Fig. 1K). More importantly, the frequency of CD13+ cells, as detected by fluorescence-activated cell sorting, was significantly increased in organoids following treatments of chemotherapeutic agents (Fig. 1L). Together, these data imply a universal chemoresistance of CD13+ CSCs in HCCs.
2. Enhancement of Tyr metabolism in CD13+CSCs from HCCs
In comparison with differentiated tumor cells, CSCs are more dependent on glycolysis [16]. Unexpectedly, CD13+ cells had negligible glycolytic activity than the matched CD13- counterpart (S1A Fig.). Glucose and glutamine are essential energy sources for cancer cells [17]. Unlike CD13- cells (S1B Fig.), however, deprivation of glucose or glutamine did not affect the viability of CD13+ fraction (Fig. 2A), suggesting a switch to alternative nutrient supply. Removing essential nutrients, one at a time, revealed significant apoptosis upon deprivation of leucine, valine, and most notably tyrosine (Fig. 2A). Deprivation of phenylalanine, a precursor of Tyr, did not affect cell survival (Fig. 2A), due to deficiency of phenylalanine hydroxylase in these cells. A significant reduction of Tyr concentration in culture medium during 24 hours incubation was observed in CD13+ but not CD13- cells (Fig. 2B). Tyr consumption by CD13+ cells was accompanied by higher expression of enzymes in Tyr degradation (tyrosine aminotransferase [TAT], 4-hydroxyphenylpyruvic acid dioxygenase [HPD], homogentisate 1,2-dioxygenase [HGD], maleylacetoacetate isomerase [MAAI], and fumarylacetoacetase [FAH]) (Fig. 2C). Furthermore, we infused 13C-Tyr into HepG2-xenografted nude mice and examined 13C label incorporation into Tyr metabolites (para-hydroxyphenylpyruvate, homogentisic acid, 4-maleylacetoacetate, 4-fumarylacetoacetate [4-FAA]) (S1C Fig.) in CD13+ cells. As shown in Fig. 2D, Tyr tracing experiments in HepG2 xeno-grafts demonstrated higher flux through Tyr degradation pathway in CD13+ than in CD13- cells.
The differential dependence on Tyr metabolism between CD13+ and CD13- cells prompts us to explore the underlying mechanism(s). We examined whether CD13 is the key determinant for activation of tyrosine metabolism in CD13+ CSCs. Notably, metabolomic analysis using 13C-Tyr tracing revealed that CD13 knockdown decreased Tyr uptake (S1D Fig.), attenuated the flux of intermediates into Tyr degradation (Fig. 2E) and increased glycolytic rate (S1E Fig.). On the contrary, overexpression of CD13 in CD13- cells recapitulated the metabolite phenotype observed in the CD13+ counterpart (Fig. 2F, S1F and S1G Fig.), highlighting the specificity of CD13 for metabolic switch in CD13+ subpopulation. Consistently, Tyr deprivation led to massive apoptosis in cultured CD13+ cells and spared > 95% CD13- fraction (Fig. 2G).
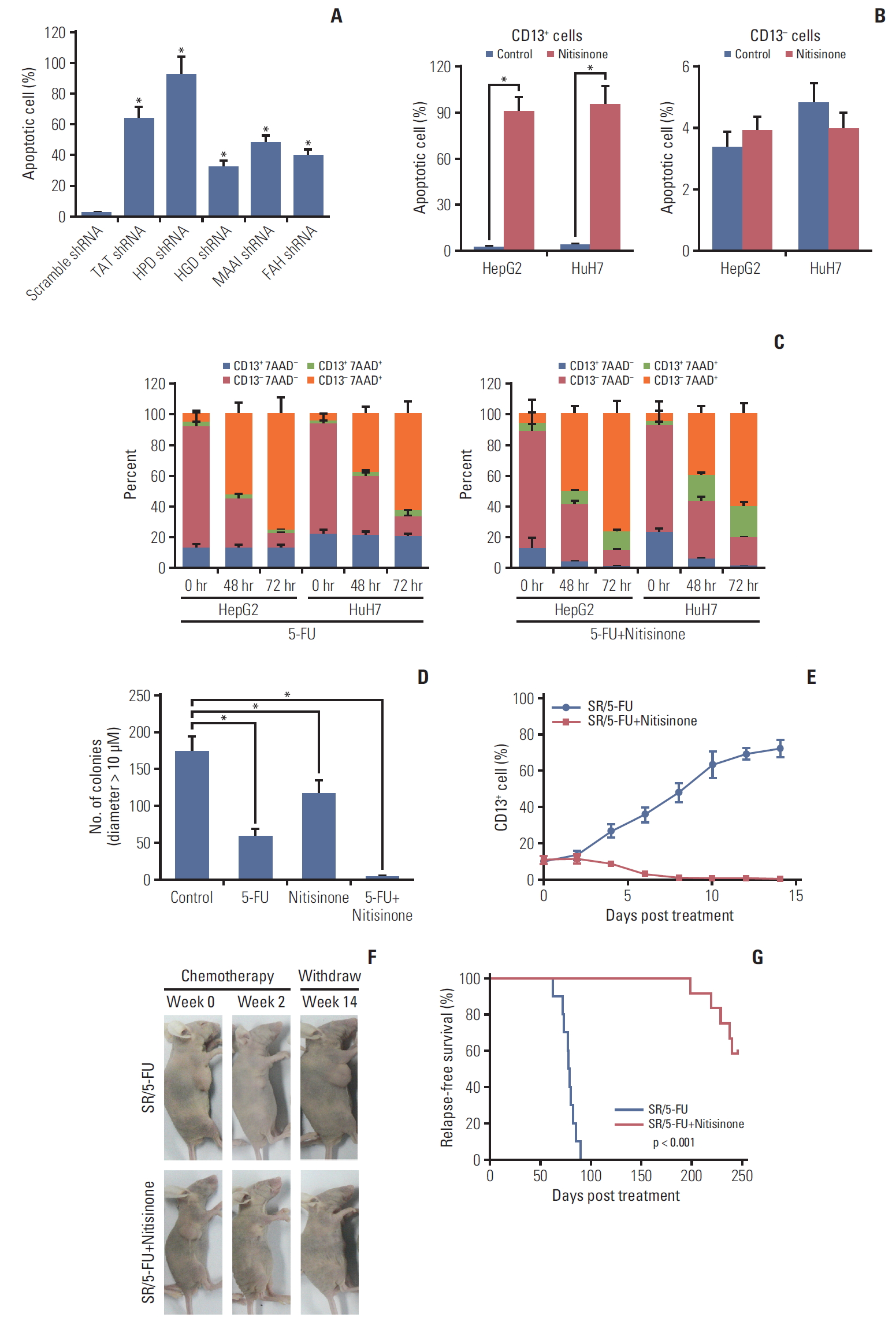
3. Ablating CD13+ CSCs and postponing recurrence in HCCs by nitisinone via targeting Tyr metabolism
Since CD13+ CSCs in HCCs are Tyr addictive, we examined whether Tyr metabolism is an effective target for preventing tumor recurrence. As almost all enzymes in Tyr degradation were found differentially regulated in CD13+ CSCs, we explored shRNAs against TAT, HPD, HGD, MAAI, and FAH in a series of preliminary cytotoxicity assays. Here, shRNA against HPD revealed the most prominent results (Fig. 3A); thus, HPD was selected for subsequent studies. Nitisinone, a commercial safe drug licensed for treatment of hereditary tyrosinaemia, can interrupt tyrosine metabolism through inhibition of HPD [18]; it was prioritized for follow-up. Firstly, we evaluated the in vitro sensitivity of CD13+ cells to nitisinone. Cytotoxicity assays confirmed a dramatic targeting of CD13+ cells with much less effect in CD13- compartment (Fig. 3B). Flow cytometric quantitation of Hep3G and HuH7 cells revealed a complete loss of CD13+ cells under nitisinone treatment, overcoming the 5-FU–induced enrichment of this fraction (Fig. 3C). The tumor-suppressive potential of ablating CD13+ CSCs was further demonstrated in 3D collagen-embedded spheroids of Hep3G cells, a model which more readily mimics the hepatic carcinoma microenvironment. Treatment of Hep3G cells with a single pulse of nitisinone plus 5-FU over 72 hours substantially reduced longterm growth in soft agar and the number of 3D colonies remained at almost undetectable levels, even after 5 weeks of culture in drug-free medium (Fig. 3D). These results indicate that even a relatively short-term treatment can yield a sustained anti-tumor effect.
Nitisinone treatment also improved the tumor-suppressive potential of chemotherapy in vivo. HepG2 cells were subcutaneously transplanted into BALB/c nude mice, and were allowed to grow until tumor reached a comparable size prior to receiving doxorubicin/5-FU regimen with or without nitisinone. Nitisinone alone gradually eliminated CD13+ cells within 8 days (Fig. 3E). Consequently, the combined treatment significantly postponed the time to recurrence (by > 21 weeks vs. mice receiving FOLFOX alone) (Fig. 3F and G).
4. Entry of Tyr-derived mitochondrial acetyl-CoA into TCA cycle
In Tyr degradation pathway, 4-FAA is catabolized by FAH to yield end-products, fumarate and acetoacetate, after which the latter is converted into acetyl-CoA (S1C Fig.). In vitro experiments using 13C-labeled Tyr showed ~53% contribution of the acetyl-CoA pool by Tyr in CD13+ cells but negligible contribution of Tyr in CD13- cells (Fig. 4A). In contrast, experiments using 13C-glucose or 13C-glutamine revealed < 20% contribution to the acetyl-CoA pool in CD13+ cells (Fig. 4A). Furthermore, targeted metabolomic analysis using 13C-labeled Tyr showed increased Tyr-derived 4-FAA (and total 4-FAA) and decreased acetyl-CoA (and total acetyl-CoA) by FAH knockdown (S2A and S2B Fig.), again suggesting Tyr contributes to the maintenance of the acetyl-CoA pool in CD13+ cells.
Acetyl-CoA from Tyr was present in both mitochondria and nucleus (Fig. 4B). Mitochondrial acetyl-CoA enters into the TCA cycle for generation of energy. TCA activity in CD13+ cells, as reflected by oxygen consumption rate (OCR), was 3.5-fold higher than in the matched CD13- cells; deprivation of Tyr, but not other essential nutrients, eliminated such a difference (Fig. 4C left, S2C Fig.). The importance of oxidative phosphorylation was further demonstrated by increased apoptosis upon treatment with the mitochondrial ATP synthase inhibitor oligomycin (Fig. 4C right). Of note, exogenous acetyl-CoA or cell-permeable dimethyl-fumarate (DMF) alone did not restore OCR upon Tyr deprivation, whereas combination of acetyl-CoA and DMF dramatically rescued OCR in CD13+ cells (Fig. 4D), indicating that Tyr yields acetyl-CoA and fumarate to support TCA cycle. In line with these, Tyr is the major carbon source for the TCA cycle in CD13+ CSCs, as determined by in vivo infusing HepG2-xenografted mice with uniformly labeled 13C-glucose, 13Cglutamine, and 13C-tyrosine, respectively (Fig. 4E).
We further determined the bioenergetic properties of CD13+ CSCs treated with nitisinone. Nitisinone inhibited homogentisate production (S2D Fig.) and induced severe impairment of OCR (Fig. 4F) in CD13+ cells. The relatively lower OCR can be compensated by upregulated glycolysis. However, these cells were not able to induce glycolysis (S2E Fig.), suggesting that nitisinone selectively inhibits energy generation in this subpopulation. Indeed, nitisinone rapidly depleted cellular ATP in CD13+ cells (Fig. 4G), whereas it had no effect in the ATP content of CD13- fraction (S2F Fig.). As nitisinone induced mitochondrial dysfunction and energy depletion in CD13+ CSCs, we asked if oxidation was promoted. Nitisinone induced a clear upregulation of mitochondrial reactive oxygen species (ROS) in CD13+ cells, accompanied with decreased levels of reduced glutathione (Fig. 4H). Therefore, HPD inhibition by nitisinone severely impaired redox balance and energy control in CD13+ CSCs, thereby eliminating this fraction.
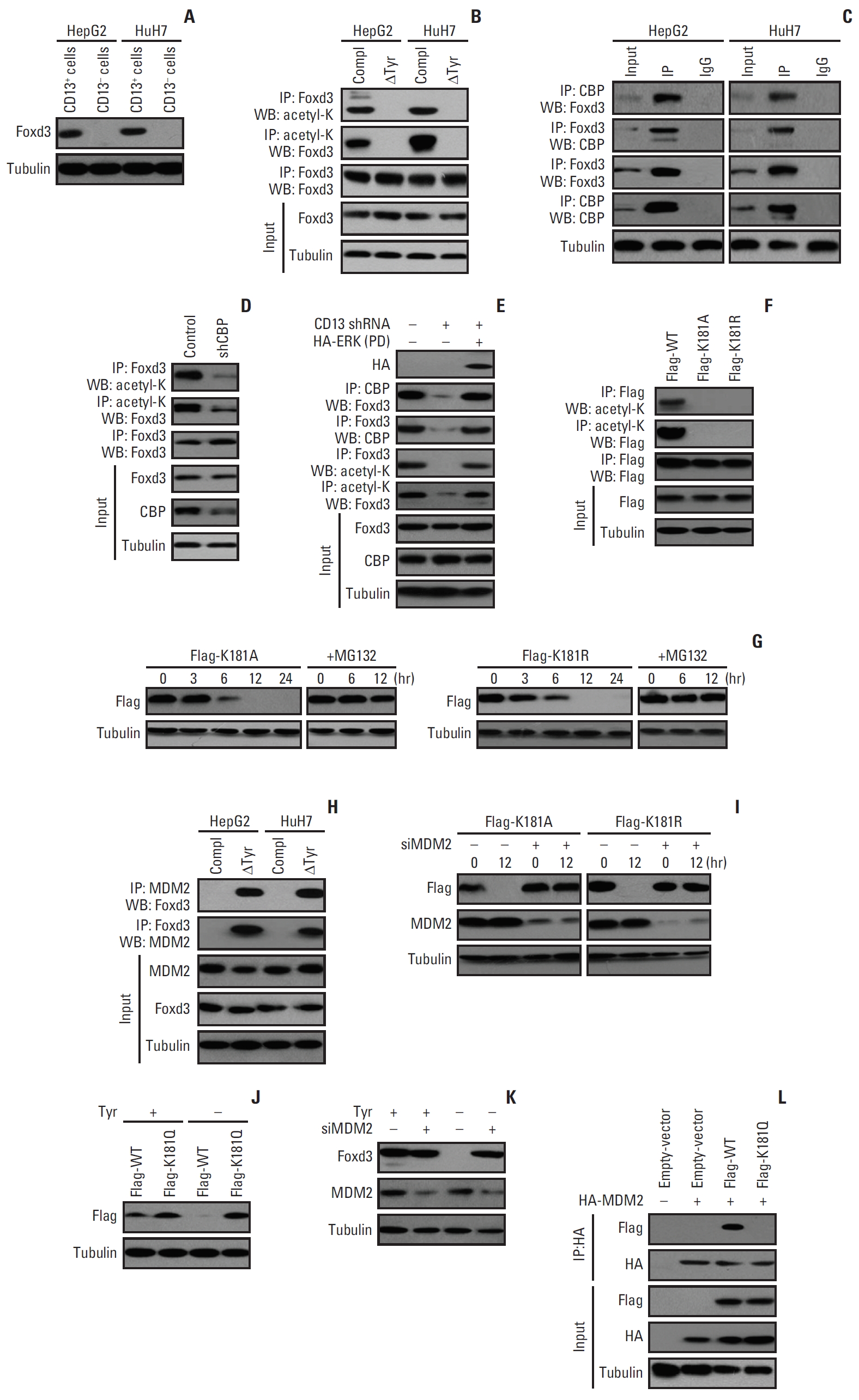
5. Prevention of Foxd3 degradation by Tyr-derived nuclear acetyl-CoA
Nanoflow liquid chromatography-mass spectrometry analysis indicated that nuclear acetyl-CoA produced by Tyr metabolism does not contribute acetyl group for histone acetylation in CD13+ cells (S3A Fig.). Spiking CD13+ cells with 13C-Tyr followed by immunoprecipitating acetylated peptides from nuclear extracts resulted in 14 nuclear proteins containing 13C-acetyllysine (S4 Table). No 13C-acetyl peptides corresponding to known acetylation sites on core histones were discovered. Notably, Foxd3 knockdown impaired the resistance of CD13+ cells to 5-FU or SR to a greater extent than with knockdown of any other candidate proteins (S3B Fig.). Foxd3 was preferentially expressed in CD13+ fraction, but not in the matched CD13- cells (Fig. 5A), at least partially explaining the therapeutic vulnerability of CD13+ cells. Therefore, we focused on Foxd3 in subsequent studies. Foxd3 acetylation was detectable in CD13+ cells (Fig. 5B). Tyr deprivation in culture medium abrogated Foxd3 acetylation (Fig. 5B). Also, Foxd3 was no longer acetylated in a TAT or FAH mutant (S3C Fig.). To identify the acetyltransferase responsible for Foxd3 acetylation, we knocked down 12 lysine acetyltransferases with siRNA (S3D Fig.), one at a time, and found that only CBP regulated Foxd3 acetylation (S3E Fig.). Immunoprecipitation further revealed association between CBP and Foxd3 (Fig. 5C). Transduction of CD13+ cells with shRNA targeting CBP abrogated Foxd3 acetylation (Fig. 5D). Previous studies reported that mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinases could increase CBP acetyltransferase activity by phosphorylating CBP at its C-terminal [19]. Since signaling via ERK1/2 lies downstream of the CD13/MAPK axis [20], treatment of CD13+ cells with CD13 shRNA decreased the level of the CBP-Foxd3 complex and blocked Foxd3 acetylation (Fig. 5E), which were largely rescued by a constitutively active ERK mutant (Fig. 5E). These data suggested that ERK1/2 signaling is required for CBP-Foxd3 interaction and Foxd3 acetylation.
Tandem mass spectrometry analysis of Flag-tagged Foxd3 identified an acetylation site at K181 (S3F Fig.). Acetylation was evident in CD13+ cells with wild-type Foxd3 (wtFoxd3) but not the acetylation-resistant K181A or K181R mutant (Fig. 5F). Incidentally, both K181 mutants were subjected to degradation and became undetectable within 12 hours of ectopic expression (Fig. 5G). Treatment with the proteasomal inhibitor MG132 increased the stability of K181 mutants (Fig. 5G), indicating that these mutants are degraded through the ubiquitin-proteosome system. ERK activation leads to FOXO proteins ubiquitination mediated by MDM2 E3 ligase [21]. We found that endogenous Foxd3 binds to MDM2 upon Tyr deprivation (Fig. 5H). After transfection of the K181A or K181R mutant, MDM2 interacted with these mutants (S3G Fig.). As expected, knockdown of MDM2 decreased turnover of K181A or K181R proteins (Fig. 5I), suggesting that K181 acetylation protects Foxd3 from MDM2-mediated degradation. In contrast, we observed undetectable level of wtFoxd3, but high level of constitutively acetylated K181Q mutant under Tyr-free conditions (Fig. 5J). MDM2 knockdown also rescued degradation of endogenous Foxd3 upon Tyr deprivation (Fig. 5K), without affecting Foxd3 mRNA (S3H Fig.), suggesting that Foxd3 expression is regulated at the posttranscriptional level. Consistently, immunoprecipitation analysis revealed an association of HA-tagged MDM2 with wtFoxd3 in HEK293T cells (due to negligible endogenous CBP activity), but not with K181Q (Fig. 5L).
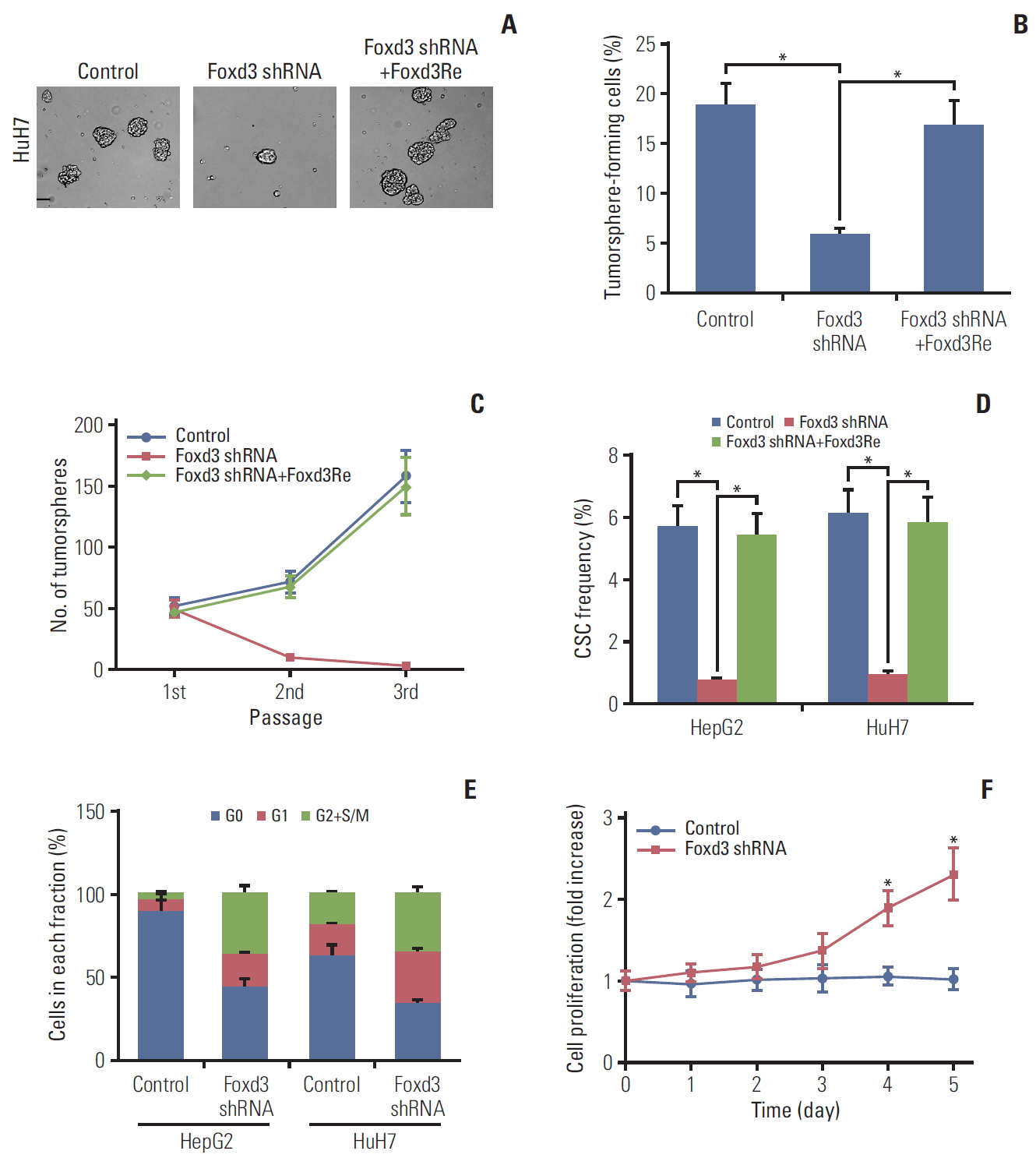
6. Impairment of self-renewal and quiescence of CD13+ cells by loss of Foxd3
Next, we examined the role of Foxd3 in the maintenance of CD13+ CSCs. In a tumorsphere experiment, Foxd3 knockdown (Foxd3KD) with an shRNA (S5A Fig.) could impair self-renewal of CD13+ cells, as reflected by significantly reduced tumorspheres formation (Fig. 6A and B) and decreased passaging and expansion (Fig. 6C). Next, we gave mice limiting doses of Foxd3KD or control CD13+ cells. Tumors generated from a single cell (with limiting dilution) were transplanted into secondary recipients. The results demonstrated that Foxd3KD decreased the frequency of self-renewing cells by > 8-folds (Fig. 6D). These effects were largely rescued by a human Foxd3 plasmid (Foxd3Re) resistant to Foxd3KD (Fig. 6A-D). Sorted CD13+ cells did not show increased propidium iodide (PI) uptake upon Foxd3KD (S5B Fig.), thereby excluding the possibility that the pool of CD13+ cells was exhausted by a process of cell death. Quiescence protects stem cells against loss of self-renewal capacities [22]. In our experiments, Hoechst/PyroninY staining revealed that Foxd-3KD in CD13+ cells decreased the percentage of cells in the G0 phase, and increased the proportion in G1/S/G2M phases (Fig. 6E). Accordingly, CD13+ cells proliferated faster after Foxd3KD (Fig. 6F). These data indicated that increased proliferation likely contributes to the reduced self-renewal of Foxd3KD cells by diminishing the pool of quiescent stem cells.
Discussion
Most tumor therapies, including chemo- or targeted-therapies and ionizing radiation, preferentially affect cycling cells but spare quiescent cells. Here we showed that functionally defined CD13+ CSCs in HCCs are characterized as quiescent and do not respond to chemotherapeutic agents. Drug sensitivity is, therefore, a functional property that varies between the heterogeneous cell types comprising HCCs. As stem cell signature expression in various tumor correlates inversely with outcome [23], we proposed that the degree of CD13 at the gene or protein level relates to the size of the clinically essential pool that can cause relapse.
One strategy towards eliminating quiescent cancer cells is to selectively target unique metabolic demands of these cells [24]. In addition to changes in the metabolism of glucose and glutamine, the role of amino acids in supporting tumor growth has also been increasingly implicated. Using in vivo negativeselection RNAi screens, Possemato et al. [25] established that the serine pathway is essential to most estrogen negative breast cancers subtypes. The apoptotic pathways engaged by amino acid shortage have not been thoroughly explored, but some reports indicate that cell death occurs via mitochondrial apoptosis, possibly mediated by the Noxa/Mcl-1 axis [26]. In this study, the quiescent phenotype of CD13+ CSCs reflected a metabolic state that is characterized by higher dependence on exogenous Tyr to survive relative to the rapidly proliferating tumor bulk population. This dependence was attributed to the presence of CD13-induced signaling cascade, which constituted a unique mechanism to regulate the metabolic switch from glycolysis to Tyr degradation. Tyrosine, a non-essential amino acid, can be degraded into acetyl-CoA and fumarate to support citric acid cycle. Thus, CD13+ CSCs depend on mitochondrial function, but not glycolysis, to meet their energy demands. As mitochondrial respiration is the major cellular process that produces ROS, our findings converge on previous reports that CD13+ CSCs have relatively higher ROS than in their CD13- counterparts [8,27].
Apart from its central role in metabolism, acetyl-CoA regulates cell functions by participating in the acetylation of a variety of proteins [28]. The current study further proved that Tyr metabolism produces acetyl-CoA in the nuclear compartment to acetylate a number of non-histone proteins, including Foxd3, a transcriptional factor belonging to the Fox family. Foxd3 is expressed in dorsal and paraxial mesoderm and neural crest, as well as inner cell mass and trophectoderm of mammalian embryonic stem cells (ESCs). Foxd3 is involved in pluripotency maintenance in human ESCs, in promoting epithelial to mesenchymal transition during neural crest specification, in regulating the balance between melanocyte and Schwann cell development, and in regulation of a set of differentiation-associated genes along with NFATc3 in ESCs [29]. In the context of mutated Ras-driven tumors, activation of ERK leads to phosphorylation and subsequent degradation of FOXO proteins in an MDM2-dependent manner [21]. However, it appears that this is not the case in CD13+ CSCs in HCCs, as reflected by the protection of Foxd3 from MDM2-induced polyubiquitination and degradation when Foxd3 was constitutively acetylated in our experiments. Knockdown of Foxd3 promoted the entry of CD13+ CSCs into cell cycle, and decreased their self-renewal. As a consequence, Foxd3-deficient populations lost features characteristic of CSCs, i.e., quiescence and self-renewal, and were susceptible to chemotherapy. In this sense, Tyr metabolism not only meets the energy requirement of CD13+ CSCs, but also is essential for the maintenance of this subpopulation. Targeting the metabolic “Achilles heel” of CD13+ CSCs by nitisinone in parallel to tumor debulking by chemotherapy may be required for optimal therapeutic specificity yielding the most durable tumor remission in HCCs. Last but not least, nitisinone is an imperfect treatment, in as much as it markedly increases circulating Tyr. Managing high Tyr by using a lower protein diet to minimize adverse effects including skin rash and corneal keratopathy will be required [30].