Introduction
Biliary tract cancer (BTC) is a relatively rare tumor, but its incidence is higher in Korea than in Western countries [1]. To date, there have been no clinically validated therapeutic targets in BTC, resulting in a poor prognosis. Consequently, there is a huge unmet medical need for new drug development in BTC.
The DNA damage response (DDR) is a multi-complex network of signaling pathways involved in DNA damage repair, cell cycle checkpoints, and apoptosis. If DNA damage is accumulated, it is likely to provoke mutations or gene aberrations [2]. Disruption of the DDR has been identified in various cancers, and defects in specific DDR pathways can be compensated by other DDR pathways to maintain survival [3,4].
The DDR plays a critical role in the maintenance of genomic integrity. Approximately 450 proteins are related to the DDR. Among the huge complicated DDR networks, DNA damage sensors such as ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia and Rad3-related (ATR), DNA-dependent protein kinase (DNA-PK), and poly(ADP-ribose) polymerase (PARP) have crucial roles in the repair of single-strand breaks and double-strand breaks (DSBs) [2,3,5]. Previous studies have shed light on the action of ATR in the DDR, and furthermore, distinctions from other sensors have been discovered [6-10]. For example, the regions of RPA-coated single stranded DNA formed by replication stress or through resection of DSBs can be recognized by ATR, which maintains genomic integrity during S or G2/M phase of the cell cycle, and is particularly important for enhancing cell survival. In contrast, ATM, another core member of the DDR, is generally activated by DSBs and is not essential for cell survival [6,9]. Targeting of DDR pathways is one of the areas for new drug development and has been explored as a potential strategy for cancer therapy [2-4,9,11].
Based on recent reports, DNA damage repair pathways, which include BAP1, MSH6, BRCA1, ATM, MLH1, and MSH2, are altered in about 20% of BTC cases [12-15]. A faulty DDR generally affects the response to chemotherapeutic drugs, and there is growing evidence that tumors with defective DNA damage repair could be sensitive to certain DDR-targeted agents [4,16]. In addition, the frequent occurrence of these mutations often leads to microsatellite instability (MSI) [4], and the MSI status has an important role in the pathogenesis of cancers [13]. Notably, previous studies showed that MSI-high was present in about 25% of BTC cases [13]. Furthermore, the occurrence of TP53 gene mutations was observed in 33.9% of BTC cases [14]. TP53 is one of the most widely found tumor suppressor genes, and acts as the guardian of genomic p53 signaling pathways that control DNA damage repair, cell cycle progression, or apoptosis [17]. Therefore, various researchers have employed TP53-deficient cancer cells in DDR-targeted therapeutic studies [18]. Taken together, BTC has the potential to respond well to DDR-targeted agents. However, the DDR-targeting strategy has not yet been tested in BTC.
In this study, we aimed to evaluate a DDR-targeting strategy using an ATR inhibitor, AZD6738 [11], as monotherapy or in combination with cytotoxic chemotherapeutic agents in BTC cells. In addition, we focused on defining biomarkers for ATR inhibition in BTC.
Materials and Methods
1. Human cell lines and reagents
Nine human BTC cell lines were utilized in this research. The SNU245, SNU308, SNU478, SNU869, SNU1079, and SNU1196 cell lines were purchased from Korean Cell Line Bank (Seoul, Korea). The HuCCT-1 and TFK-1 cell lines were obtained from RIKEN BioResource Center (Ibaraki, Japan). A patient-derived cell line, SNU2670, was successfully established [19]. All cells were cultured in RPMI-1640 medium (Welgene Inc., Gyeongsan, Korea) containing 10% fetal bovine serum and 10 μg/mL gentamicin at 37°C incubator under 5% CO2. The ATR inhibitor AZD6738 was kindly provided by AstraZeneca (Macclesfield, Cheshire, UK). Gemcitabine, cisplatin, and 5-fluorouracil (5-FU) were purchased from Lilly Korea Co. (Seoul, Korea), JW Pharmaceutical Co. (Seoul, Korea), and Ildong Pharmaceutical Co. (Seoul, Korea), respectively.
2. Cell viability assay
Cells were seeded in 96-well plates and incubated overnight at 37°C. The cells were exposed to increasing concentrations of AZD6738 alone or in combination with cytotoxic agents for 5 days. Subsequently, 50 μL of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Sigma-Aldrich, St. Louis, MO) was added to each well and incubated at 37°C for 4 hours. The medium was removed and 150 μL of dimethyl sulfoxide was added to each well. The cell viability was measured at 540 nm with a VersaMax Microplate Reader (Molecular Devices, Sunnyvale, CA). The experiments were performed in triplicate.
3. Colony-forming assay
Cells were seeded in 6-well plates and exposed to various concentrations of AZD6738 or cytotoxic agents. After 10 days, the colonies were stained with Coomassie blue for 2 hours and counted using Gel Doc system software (Bio-Rad, Hercules, CA). Each experiment was repeated three times.
4. Western blot analysis
Cells were seeded in 60-mm dishes and treated with AZD6738, cytotoxic agents, or AZD6738 plus cytotoxic agents for 5 days. The cells were harvested and lysed in RIPA buffer containing protease inhibitors on ice for 30 minutes. The proteins were extracted and equal amounts of proteins were applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by western blot analyses. Primary anti-bodies against the following molecules were purchased from Cell Signaling Technology (Beverley, MA): p53 (#9282); ATR (#2790); phosphorylated ATR-Ser428 (#2853); Chk1 (#2360); phosphorylated Chk1-Ser345 (#2341); phosphorylated AKT-Ser473 (#9271); AKT (#9272); phosphorylated ERK-Thr202/Tyr204 (#9101); ERK (#9102); PARP (#9532); caspase-7 (#9492); phosphorylated CDC2 (#9111); CDC2 (#9112); and p21 (#2947). α-Tubulin (#T5168) and β-actin antibodies were purchased from Sigma-Aldrich. Anti-ATM antibody (#ab78) was obtained from Abcam Bioscience (Cambridge, UK). Anti-γH2AX antibody (#05-636) was bought from Millipore (Billerica, MA). Anti-permeability glycoprotein (p-glycoprotein) and the secondary antibodies were purchased from Thermo Scientific (Waltham, MA). The intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
5. Cell cycle analysis
Cells were seeded in 60-mm dishes and treated with various concentrations of AZD6738 for 5 days. The cells were harvested and fixed with 70% ethanol at −20°C. After 2 days, 7 μL of RNase A (20 mg/mL, Invitrogen, Carlsbad, CA) was added to each well and incubated for 10 minutes at 37°C. The cells were stained with 13 μL of propidium iodide (Sigma-Aldrich) and analyzed in a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Each experiment was repeated three times.
6. Alkaline comet assay
Cells were exposed at 0.5 μmol/L AZD6738, 0.5 μmol/L cisplatin, or their combination for 5 days. The cells were harvested, resuspended at 1×105 cells/mL in ice-cold phosphate-buffered saline (PBS), and combined with molten LMA-garose at a ratio of 1:10. Next, the samples were slightly moved onto comet slides and incubated at 4°C in the dark for 40 minutes. After immersion in precooled lysis solution at 4°C for 40 minutes, the slides were placed in freshly prepared alkaline unwinding solution (200 mM NaOH, 1 mM EDTA, pH > 13) for 30 minutes at room temperature in the dark. Electrophoresis was conducted for 30 minutes, and the samples were dried at room temperature overnight. Next, 100 μL of diluted SYBR Green staining solution was placed onto each circle of agarose, and the samples were covered with a coverslip. Tail moment and intensity were measured using the Comet Assay IV program (Andor Technology, Belfast, UK). Three independent experiments were analyzed for each condition.
7. In vivo experiments
Twenty female athymic nude mice of 4-week-old were purchased from Central Lab Animal Inc. (Seoul, Korea). SNU478 xenograft model mice were established by subcutaneous inoculation of 2×107 cells in 100 μL of PBS. When the tumor volume reached 200 mm3, the mice were randomly divided into four groups of five mice. AZD6738 was administered orally once a day at 25 mg/kg for 3 weeks and the control group was treated with vehicle (2-hydroxypropyl-β-cyclodextrin solution) by oral gavage. Cisplatin was injected intraperitoneally twice in the first week and then once weekly for next 2 weeks. Body weights and tumor sizes were measured every other day. The tumor volume was calculated using the formula: tumor volume=[(width)2×height]/2. None of the mice was killed during experiment by mistake and all mice were sacrificed finally.
8. Immunohistochemistry
Sections of paraffin-embedded xenograft tumor tissues were deparaffinized and dehydrated. Immunohistochemistry (IHC) detection of proliferating cells was conducted using an anti-Ki-67 antibody (GeneTex Inc., Irvine, CA) at a dilution of 1:100. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assays were conducted for IHC detection of apoptosis using an ApopTag In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer’s protocol. Phosphorylated Chk1 (#2341) was used at dilution of 1:50.
9. Statistical analysis
Statistical analyses were conducted using SigmaPlot ver. 10.0 (Systat Software Inc., San Jose, CA). Experimental data are presented as mean±standard error (SE). All statistical tests were two-sided. Differences were considered statistically significant for values of p < 0.01. The half-maximal inhibitory concentration (IC50) of agents was also analyzed using the SigmaPlot software. The combination effects of agents were analyzed by the combination index (CI) by CalcuSyn software, the methods described in the Chou and Talalay median effect principle were used, with CI values of < 1, 1, and > 1 indicating synergistic, additive, and antagonistic effects, respectively.
10. Ethical statement
Animal experiments were performed at the Biomedical Center for Animal Resource Development of Seoul National University (Seoul, Korea) according to institutional guidelines, with prior approval from the institutional animal care and use committee (approved number: SNU-130802-3-3).
Results
1. AZD6738 inhibits proliferation of BTC cells
Given our understanding on the roles of ATR, we hypothesized that ATM-deficient BTC cell lines may be sensitive to AZD6738. Before testing the reagent, we probed the basal ATR, ATM and p53 expression levels in the nine BTC cell lines. As shown in Fig. 1A, the ATM levels were varied. Compared with SNU308, SNU1079, SNU1196, and HuCCT-1, the other five cell lines (SNU245, SNU478, SNU869, TFK-1, and SNU2670) exhibited low ATM expression. Currently, there was no information about ATM mutation or MSI-status in these BTC cell lines. High expression of p53 was observed in SNU245, SNU308, SNU1196, HuCCT-1, and SNU2670 cells. SNU478 and SNU869 cells showed low levels of p53 expression.
To examine whether ATM or p53 levels affected the sensitivity to AZD6738, cells were exposed to increasing doses of AZD6738, and detected for their cell viability by MTT assays after 5 days of treatment. AZD6738 as a single agent strongly suppressed cell growth in the BTC cell lines (Fig. 1B). Specifically, SNU478 (IC50, 0.46 μmol/L) and SNU869 (IC50, 0.44 μmol/L), which expressed low levels of both ATM and p53, were highly sensitive to AZD6738, whereas the more resistant cell lines, such as SNU2670 (IC50, > 10 μmol/L), exhibited low expression of ATM and high expression of p53 (Table 1). To better understand the anti-proliferative effect of AZD6738, colony-forming assays were conducted. The number of colonies was markedly reduced by AZD6738 treatment for 10 days in all BTC cell lines. The IC50 values revealed that, compared with the SNU245 (> 1 μmol/L) and SNU2670 (0.9 μmol/L) cell lines, the efficacy of AZD6738 were more obviously observed in the SNU478 (0.1 μmol/L) and SNU869 (0.13 μmol/L) cell lines (Table 1).
Based on the AZD6738 sensitivity, we selected four cell lines (two sensitive cell lines, SNU478 and SNU869; two resistant cell lines, SNU245 and SNU2670) to detect related signaling pathways (Fig. 1C). Following treatment with AZD6738, ATR, phosphorylated Chk1, and p-glycoprotein were significantly reduced in the sensitive cell lines (SNU478 and SNU869). In contrast, the resistant cell lines (SNU245 and SNU2670) showed no changes in these signals. Notably, prominent dose-dependent upregulation of γH2AX was observed in the SNU478, SNU869 and SNU245 cell lines after AZD6738 treatment.
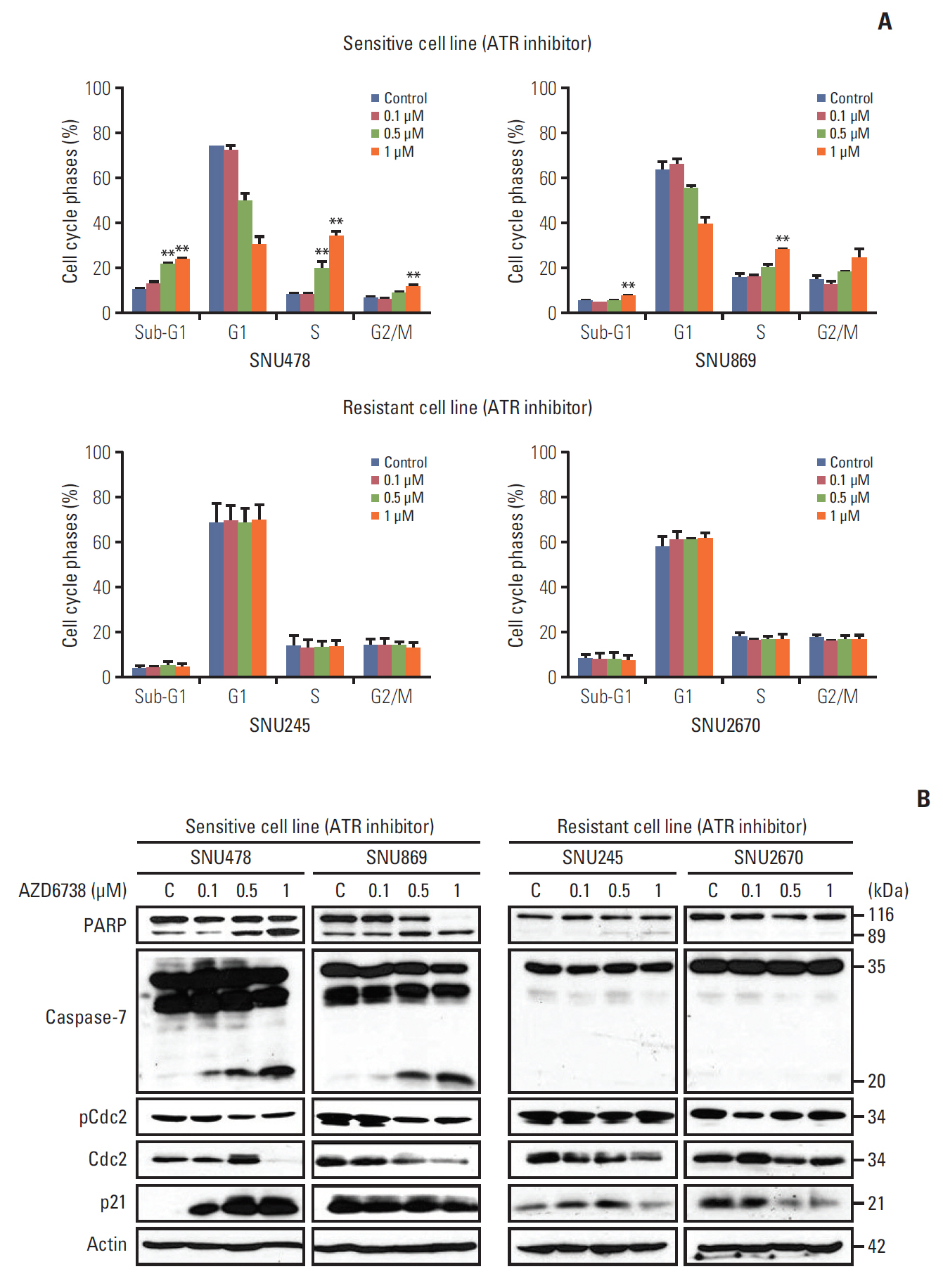
2. AZD6738 leads to cell cycle arrest at sub-G1 and G2/M phases
To assess whether AZD6738 as a single agent facilitates cell cycle arrest or apoptosis, we conducted fluorescence-activated cell sorting analyses after 3 days of treatment. The proportion of cells in S and G2/M phase as well as in sub-G1 phase escalated dose-dependently in the sensitive cell lines (SNU478 and SNU869, p < 0.01) (Fig. 2A).
In the resistant cell lines (SNU245 and SNU2670), with the exception of induction of S phase arrest in the SNU245 cell line by AZD6738 treatment, ATR inhibition did not perturb the cell cycle. These results were consistent with the signal changes observed for cell cycle progression (Fig. 2B). As expected, AZD6738 induced dose-dependent PARP and caspase-7 cleavage in the SNU478 and SNU869 cell lines (Fig. 2B). Moreover, CDC2 was reduced in both sensitive cell lines; however, increased p21 was only detected in the SNU478 cell line. In contrast, cleaved PARP and reduced CDC2 induced by AZD6738 were observed in the SNU245 cell line, but not in the SNU2670 cell line. There was no change in p-CDC2 level in both of SNU245 and SNU2670 cells. In the resistant cell lines, AZD6738 did not affect caspase-7 cleavage, but decreased p21 expression in both of them.
3. The synergistic anti-proliferative effects was observed by combined with AZD6738 and cytotoxic agents
To evaluate the synergistic effects between AZD6738 and cytotoxic agents such as cisplatin, gemcitabine, and 5-FU, the methods described in the Chou and Talalay median effect principle were used. The results of MTT assays revealed that AZD6738 exhibited synergistic anti-proliferative effects in combination with cisplatin or 5-FU in the SNU478 cell line (CI < 1). In the resistant cell lines (SNU245 and SNU2670), combination treatment of AZD6738 with cisplatin, gemcitabine, or 5-FU showed strong synergistic anti-proliferative effects compared with AZD6738 monotherapy (CI < 1) (Fig. 3A). Similar results were obtained in colony-forming assays. There was no synergistic effect between AZD6738 and cisplatin for the SNU869 cell line (Fig. 3B). Consistent with the MTT assay results AZD6738 exerted synergistic anti-proliferative effects in the SNU478 and SNU2670 cell lines when co-treated with cisplatin (p < 0.01). Although we did not observe synergistic effects on the SNU245 cell line in colonyforming assays, the proliferation of the cells treated with combination therapy was obviously inhibited compared with AZD6738 monotherapy (p < 0.01).
We confirmed the above results by detecting the primary related signals. As shown in Fig. 3C, phosphorylated AKT, phosphorylated ERK and total PARP were more downregulated in SNU478 and SNU2670 cell lines when AZD6738 (1 μmol/L) was combined with cisplatin (1 μmol/L) compared to each drug alone. Notably, the combination treatment synergistic downregulated p-Chk1 level in SNU478 cells. The combination regimens also resulted to decreased total and increased cleaved caspase-7 in SNU478 and SNU2670 cells, respectively. Although phosphorylated AKT and phosphorylated ERK were not blocked by combination treatment in SNU245 cells, we observed increased cleavage of PARP and caspase-7 than mono-treatment. Blocked phosphorylated AKT, and phosphorylated ERK, or augmented caspase-7 cleavage were not found by co-treated group in SNU869 cells, there was only exhibited decreased total PARP expression. All of these results support that AZD6738 combined with cisplatin had prominent synergistic effects to combat BTC cells.
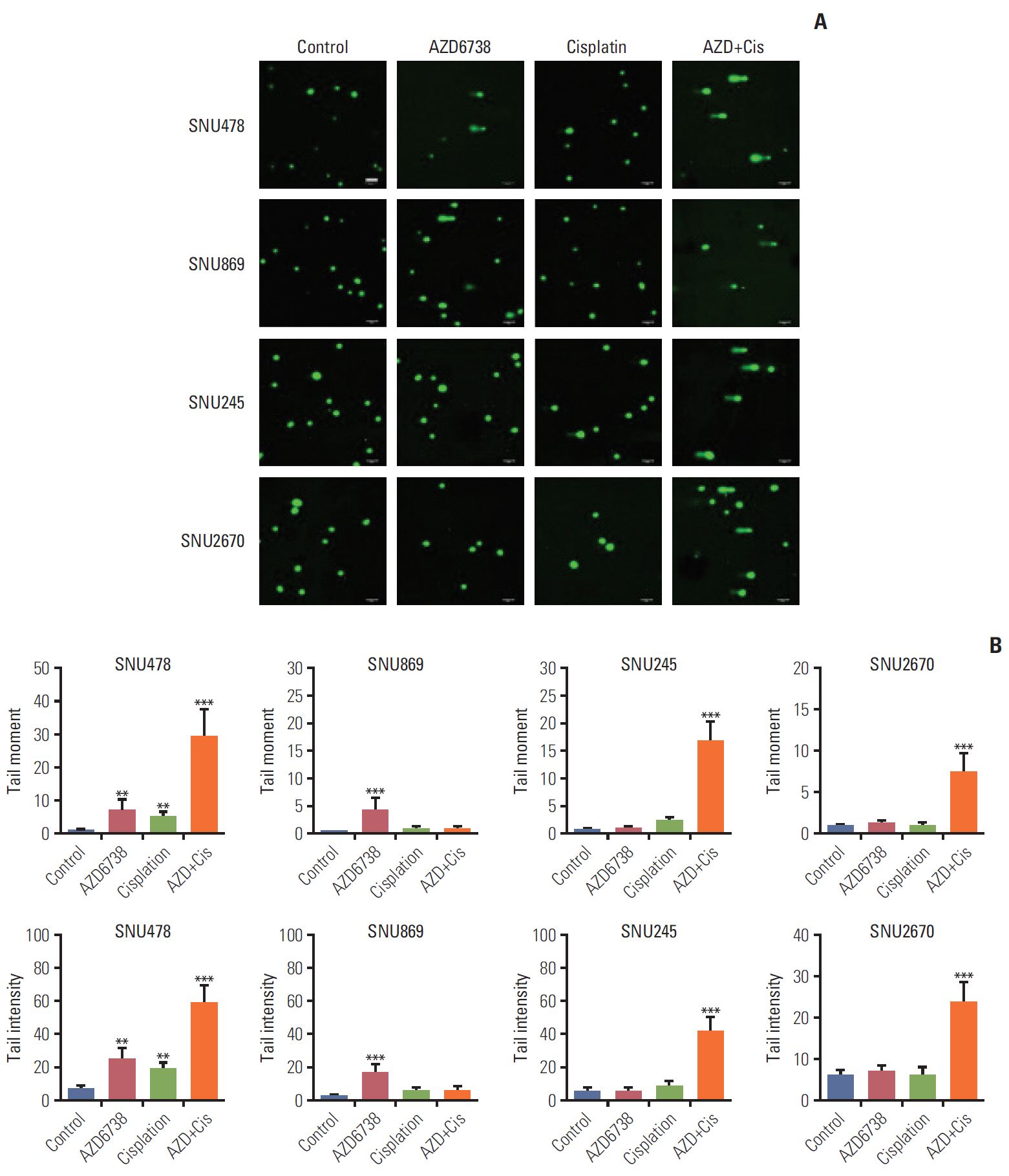
4. Co-treatment of AZD6738 and cisplatin strongly induces DNA strand breaks
ATR is known to be responsible for suppressing DNA break formation and DSB DNA repair to solve DNA damage [9]. We conducted alkaline comet assays to detect DNA damage by AZD6738. Accumulation of DNA damage was found in the SNU478 and SNU869 cell lines after treatment with AZD6738 alone, while AZD6738 (0.5 μmol/L) had insufficient capacity to induce DNA damage in the SNU245 and SNU2670 cell lines. In addition, when the cells were exposed to both AZD6738 and cisplatin, accumulation of DNA damage was observed not only in the SNU478 cell line, but also in the SNU245 and SNU2670 cell lines (Fig. 4A). The analyses of tail moment and intensity supported our observations (Fig. 4B). These results indicated that AZD6738 has a prominent effect on DNA strand breaks and further enhances cisplatin efficacy.
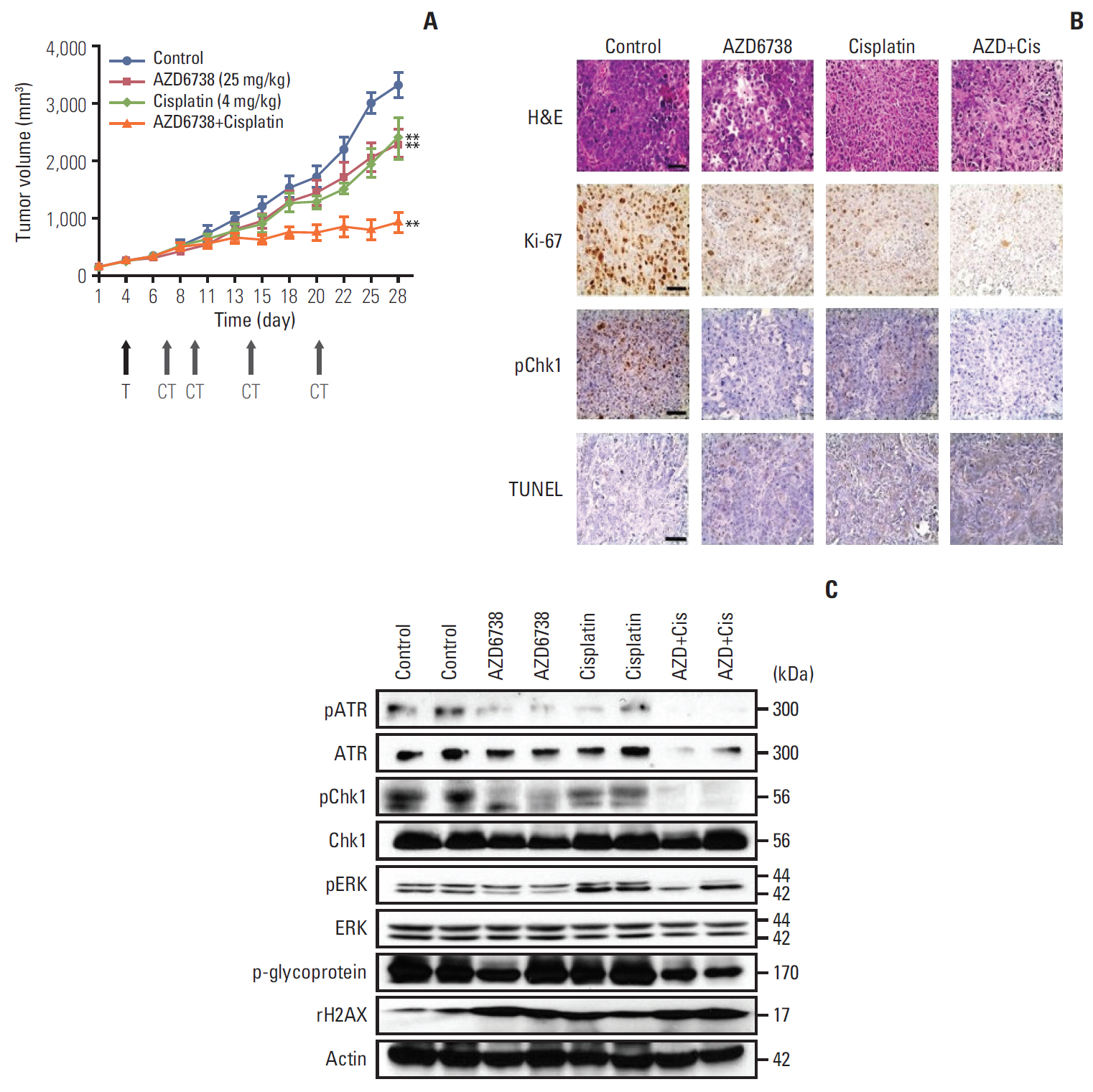
5. AZD6738 shows potent anti-tumor effects in vivo as a monotherapy and in combination with cisplatin
To further evaluate the anti-tumor effect of AZD6738, we employed SNU478 xenograft model mice. AZD6738 monotherapy (25 mg/kg) suppressed the tumor growth compared with the control group (p < 0.01) (Fig. 5A). In particular, the combination of AZD6738 with cisplatin significantly repressed tumor growth compared with monotherapy (p < 0.01). As a marker of cell proliferation, Ki-67 was reduced by AZD6738 or cisplatin and further decreased by their combination treatment in the tumor tissues (Fig. 5B). Phosphorylation of Chk1 was reduced by AZD6738 monotherapy and more profoundly decreased by the combination treatment. Moreover, the combination of AZD6738 with cisplatin upregulated TUNEL expression. Next, proteins were harvested from two different isolated tumor tissues in each group for western blotting analyses (Fig. 5C). Phosphorylated ATR, phosphorylated Chk1, phosphorylated ERK, and p-glycoprotein were more strongly blocked by the combination treatment than by AZD6738 or cisplatin alone. γH2AX as a DNA damage marker was increased in the AZD6738 alone, cisplatin alone, and combination groups compared with the control group.
Discussion
The potential efficacy of ATR inhibition for targeting the DDR has been identified in different types of cancers [8,18,20]. Currently, there are several ATR inhibitors under development. Particularly, AZD6738, as an orally bioavailable the serine/threonine protein kinase ATR inhibitor with more excellent selectivity for ATR inhibition and pharmacokinetic characteristics compared with the others [10]. In addition, AZD6738 has been tested in several cancers including leukemia, gastric and ovarian cancers [20-22]. In the case of BTC, the DDR-targeting strategy toward the development of new drugs has not yet been tested. In this study, we reveal the therapeutic efficacy of this ATR inhibitor in BTC cells and provide basic evidence for further studies on the DDR-targeting strategy for new drug development in BTC.
In recent years, inhibitors of ATR have been studied by some researchers, and cell sensitivity to these ATR inhibitors has been determined [20,23-25]. Tumor types with loss of ATM function or high replication stress are expected to be more susceptible to ATR inhibition [18]. In breast cancer cells with ERCC1 deficiency, ATR pathway inhibition was synthetically lethal [25]. Meanwhile, some studies identified a series of predictive biomarkers that affect sensitivity to ATR inhibition, including TP53 or ARID1A deficiency and CDC-25A overexpression [18,23,24].
According to previous publications, ATM-low or p53-deficient cancer cells are considered strong candidates for DDR-targeted therapy [18,21]. In our study, we found that the SNU478 and SNU869 cell lines, which were the most sensitive to AZD6738, exhibited low expression of both ATM and p53 among the nine BTC cell lines examined. Meanwhile, the SNU245 and SNU2670 cell lines, which were resistant to AZD6738, expressed a low level of ATM and high level of p53. These observations indicate that the levels of both ATM and p53 may contribute to the cellular response to ATR inhibition. Among the nine BTC cell lines, four cell lines (SNU478, SNU869, SNU1196, and HuCCT-1) harbored TP53 mutations (Broad Institute Cancer Cell Line Encyclopedia). Despite the presence of TP53 mutations in these cell lines, the low level of p53 protein expression was more crucial factor for their response to AZD6738 (Fig. 1A).
ATR signaling pathways broadly mediate cell proliferation, cell cycle progression, apoptosis, and DNA damage repair [9,10]. In our study, we found that AZD6738 reduced Chk1 phosphorylation in both the SNU478 and SNU869 cell lines. Consistent with our findings, a recent report described that an ATR inhibitor induced decreased Chk1 phosphorylation in sensitive cells, but not in resistant cells [20]. Chk1 plays a critical role in the DNA replication stress response or DNA damage repair. Chk1 is not only mediated by ATR, but also controlled by ATM or DNA-PK [2,7]. Therefore, it is meaningful to downregulate phosphorylated Chk1 using AZD6738.
We also found that AZD6738 reduced p-glycoprotein in both sensitive cell lines (SNU478 and SNU869). Glycoproteins are proteins located on the cell surface membrane that regulate drug efflux to affect drug resistance [26]. Consistent with our findings, other articles clarified that inhibition of ATR-Chk1 signaling effectively reduced the levels of p-glycoprotein [27].
ATR is known to activate S phase and G2/M checkpoint to rescue cell death [20]. In our study, AZD6738 induced G2/M arrest and sub-G1 phase increase through accumulation of PARP and caspase-7 cleavage in the sensitive cell lines (SNU478 and SNU869). Moreover, given our understanding of CDK, decreased CDC2 (also known as CDK1) contributed to the cell cycle arrest [28]. A decrease in CDC2 by AZD6738 was observed in the sensitive cell lines (SNU478 and SNU-869) and a resistant cell line (SNU245).
Growing evidence suggests that an ATR inhibitor combined with cisplatin can synergistically inhibit tumor growth in different types of cancer [20,27,29]. We also evaluated this combination treatment using proliferation assays, alkaline comet assays, and xenograft studies. As a consequence, we identified significant synergistic effects in the SNU478, SNU245, and SNU2670 cell lines. These findings were not surprising, because cisplatin is cytotoxic through its binding to DNA and blockade of replication, leading to DSBs [8]. Early studies demonstrated that ATR inhibition was synergistic with cisplatin and could overcome resistance to cisplatin [18,20,27,29]. In ATM-deficient non-small cell lung cancer, AZD6738 plus cisplatin promoted accumulation of cells at the G1/S border and in early S phase [30]. However, in our study, we observed that the combination therapy induced G2/M or S phase arrest in the SNU869 and SNU2670 cell lines, while the percentage of sub-G1 phase was increased in the SNU478 and SNU869 cell lines (data not shown). In urothelial bladder cancer, ATR-Chk1 inhibition increased cisplatin-adduct DNA levels and co-treatment inhibited p-glycoprotein expression [27]. Similarly, we detected that p-glycoprotein was downregulated in the xenograft combination group. The synergistic effect of AZD6738 combined with cisplatin was very encouraging.
Our SNU478 xenograft model mice presented consistent findings with our in vitro results. We observed that AZD6738 or cisplatin monotherapy each suppressed tumor growth efficiently in vivo. These findings were consistent with recent reports describing that ATR inhibitor monotherapy can disrupt tumor growth by inhibiting the DDR [8,9]. It is noteworthy that the combination therapy provided more efficient inhibition than the monotherapy by significantly blocking Chk1 phosphorylation, p-glycoprotein, and γH2AX accumulation.
Taken together, the DDR-targeting strategy with an ATR inhibitor demonstrated antitumor activity alone or in combination with cytotoxic chemotherapeutic agents in BTC. This supports further clinical development of the DDR-targeting strategy in BTC patients. Our preclinical data could provide new prospects for BTC patients and establish a basis for further research.