A Phase 1b/2a Study of GC1118 with 5-Fluorouracil, Leucovorin and Irinotecan (FOLFIRI) in Patients with Recurrent or Metastatic Colorectal Cancer
Article information

Abstract
Purpose
GC1118 is a novel antibody targeting epidermal growth factor receptor (EGFR) with enhanced blocking activity against both low- and high-affinity EGFR ligands. A phase 1b/2a study was conducted to determine a recommended phase 2 dose (RP2D) of GC1118 in combination with 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI) (phase 1b) and to assess the safety and efficacy of GC1118 plus FOLFIRI as a second-line therapy for recurrent/metastatic colorectal cancer (CRC) (phase 2a).
Materials and Methods
Phase 1b was designed as a standard 3+3 dose-escalation study with a starting dose of GC1118 (3 mg/kg/week) in combination with biweekly FOLFIRI (irinotecan 180 mg/m2; leucovorin 400 mg/m2; 5-fluorouracil 400 mg/m2 bolus and 2,400 mg/m2 infusion over 46 hours) in patients with solid tumors refractory to standard treatments. The subsequent phase 2a part was conducted with objective response rate (ORR) as a primary endpoint. Patients with KRAS/NRAS/BRAF wild-type, EGFR-positive, recurrent/metastatic CRC resistant to the first-line treatment were enrolled in the phase 2a study.
Results
RP2D of GC1118 was determined to be 3 mg/kg/wk in the phase 1b study (n=7). Common adverse drug reactions (ADRs) observed in the phase 2a study (n=24) were acneiform rash (95.8%), dry skin (66.7%), paronychia (58.3%), and stomatitis (50.0%). The most common ADR of ≥ grade 3 was neutropenia (33.3%). ORR was 42.5% (95% confidence interval [CI], 23.5 to 62.0), and median progression-free survival was 6.7 months (95% CI, 4.0-8.0).
Conclusion
GC1118 administered weekly at 3 mg/kg in combination with FOLFIRI appears as an effective and safe treatment option in recurrent/metastatic CRC.
Introduction
Colorectal cancer (CRC) is the third most common solid malignancy and the second leading cause of cancer-related death, with the global number of new cases and deaths per year approximating 1.8 million and 900,000, respectively [1]. It is estimated that approximately 50% of CRC patients will eventually develop metastases [2]. The prognosis in such cases is grave, with fewer than 20% of patients surviving more than 5 years from the diagnosis of metastasis [3].
Epidermal growth factor receptor (EGFR) is a cell membrane growth factor receptor playing a key role in the pathogenesis of various malignancies including CRC [4,5]. Anti-EGFR monoclonal antibodies are a group of drugs that bind to the extracellular domain of EGFR, competing with endogenous ligands of the receptor to inhibit the activation of EGFR tyrosine kinase [6]. Anti-EGFR antibodies, namely cetuximab and panitumumab, in combination with chemotherapy, can improve survival in RAS wild-type metastatic CRC and are the standard treatment in this subset of patients [3]. Unfortunately, innate or acquired resistance following the treatment is an issue that still needs to be addressed [7,8].
GC1118 (GC Biopharma Corp., Yongin, Korea) is a novel monoclonal anti-EGFR antibody binding to a distinct epitope from those bound by cetuximab and panitumumab [9]. Unlike the latter two agents, GC1118 shows enhanced blocking activity not only to low- but also to high-affinity EGFR ligands [9]. Previous preclinical studies using cell lines, animal models, and patient-derived xenografts demonstrated that GC1118 can produce a stronger inhibitory effect than other anti-EGFR antibodies. Moreover, GC1118 also showed activity against cancer cells that developed resistance to other anti-EGFR monoclonal antibodies [10-13]. The preliminary clinical data on the efficacy and tolerability of GC1118 monotherapy were recently confirmed in a first-in-human phase 1 study involving patients with advanced CRC and gastric cancer [14].
The aims of this study were to determine the maximum tolerated dose (MTD) and the recommended phase 2 dose (RP2D) of GC1118 when administered in combination with 5-fluorouracil (5-FU), leucovorin, and irinotecan (FOLFIRI) (phase 1b) and to assess the safety and efficacy of GC1118 in combination with FOLFIRI as a second-line therapy for recurrent/metastatic CRC (phase 2a).
Materials and Methods
1. Study design and patients
This open-label, non-randomized study was conducted in two phases (Fig. 1). The first phase (phase 1b) was designed as a standard 3+3 dose-escalation study with the primary purpose of finding the RP2D of GC1118, administered together with FOLFIRI. The starting dose of GC1118 was 3 mg/kg, and the decision to increase the dose to 4 mg/kg or determine an RP2D below 4 mg/kg was to be made by the data monitoring committee (DMC). After RP2D establishment, the study proceeded to the second phase (phase 2a), which was designed according to Simon’s optimal two-stage design [15] with objective response rate (ORR) as a primary endpoint.
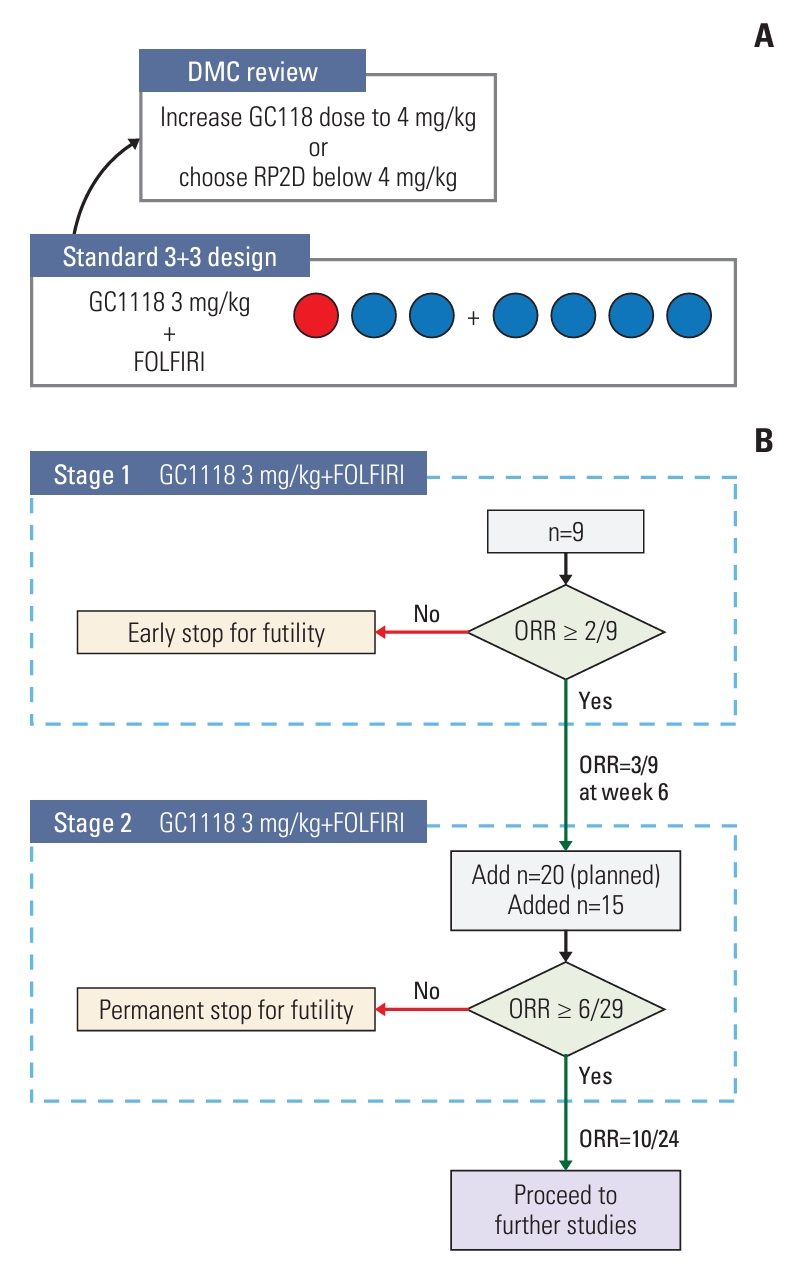
Study scheme. (A) The dose escalation phase of the phase 1b study followed a 3+3 design. A red circle indicates a patient with dose-limiting toxicity, which was grade 4 neutropenia that lasted ≥ 7 days. (B) Phase 2a study was conducted following Simon’s optimal two-stage design. DMC, data monitoring committee; FOLFIRI, 5-fluorouracil, leucovorin, and irinotecan; ORR, objective response rate; RP2D, recommended phase 2 dose.
Patients with recurrent or metastatic solid tumors refractory to the standard treatment were eligible for the phase 1b study. As for the phase 2a study, patients with KRAS/NRAS/BRAF wild-type, EGFR-positive (confirmed via immunohistochemistry), recurrent or metastatic CRC who had failed the first-line treatment containing fluoropyrimidine with or without oxaliplatin were enrolled. All patients in the phase 2a study had to have at least one measurable target lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST 1.1). Other eligibility criteria applied to both study phases included the following conditions: age ≥ 19 years; Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; life expectancy ≥ 3 months; adequate bone marrow function (absolute neutrophil count ≥ 1,500/μL, hemoglobin ≥ 9.0 g/dL, platelet ≥ 105/μL); serum creatinine ≤ 1.5 times upper limit of normal (ULN); total bilirubin ≤ 2.0 mg/dL; and aspartate aminotransferase and alanine aminotransferase ≤ 3 times ULN (up to 5 times ULN in case of hepatic metastasis). Patients with the following conditions were excluded from the study: other cancer history (exceptions included non-melanoma skin cancer that had been completely resected and disease-free for ≥ 3 years, completely resected carcinoma in situ or superficial bladder cancer, etc.); contraindication to GC1118 or FOLFIRI; use of systemic chemotherapy, immunotherapy, hormone therapy, or radiotherapy within 21 days before enrollment; brain metastasis; active infection requiring systemic antimicrobial treatment; chronic inflammatory bowel disease; or interstitial lung disease or pulmonary fibrosis. In addition, for patients to be enrolled in the phase 2a study, previous treatment with anti-EGFR antibodies or irinotecan was an additional exclusion criterion.
The study protocol was approved by the institutional review boards of participating study centers, and all patients provided written informed consent before any study-related procedures were carried out. The study was prospectively registered (Clinicaltrials.gov NCT03454620).
2. Treatment
In both 1b and 2 phases, each cycle of the study treatment consisted of two weekly administrations of GC1118 and one biweekly administration of FOLFIRI. GC1118 was given via intravenous infusion for 60 minutes. On the first day of each cycle when GC1118 and FOLFIRI were given together, GC1118 was administered first, followed by FOLFIRI after a 60-minute break. FOLFIRI consisted of simultaneous intravenous infusions of irinotecan 180 mg/m2 and leucovorin 400 mg/m2 over 120 minutes (sequential infusions of irinotecan for 90 minutes and leucovorin for 120 minutes were also allowed) followed by a bolus intravenous injection of 5-FU 400 mg/m2 and a continuous infusion of 5-FU 2,400 mg/m2 over 46 hours. The dosages of GC1118, 5-FU, or irinotecan were reduced, delayed, or discontinued in response to the toxicities related to the study treatment, according to the dosage modification guidelines (S1 and S2 Tables).
3. Study endpoints and procedures
A MTD was defined as the highest dose at which a dose-limiting toxicity (DLT) occurred at a frequency of 1/6 or less. The definition of DLT is provided in the S3 Table. The DMC determined the RP2D based on the DLTs and observed overall toxicities. Any treatment-emergent adverse events (TEAEs), regardless of causality, were collected throughout the study period. Any abnormality in physical examination, clinical laboratory results, echocardiography, or electrocardiography detected after initiation of study treatment was also reported as a TEAE.
Efficacy outcomes included ORR, defined as a proportion of patients achieving complete response (CR) or partial response (PR); disease control rate (DCR), defined as a proportion of patients achieving CR or PR or maintaining stable disease; progression-free survival (PFS); overall survival (OS); time to respond; duration of response; and percent change in the sum of the longest diameters of target lesions from baseline. PFS was defined as the time from the first dosing of GC1118 to the date of the radiological confirmation of progressive disease (PD) or death due to any cause. Patients without PD at the end of the study were censored at the date of the last tumor evaluation. OS was defined as the time from the first dosing of GC1118 to the date of death from any cause or the date of the last follow-up. Patients without the confirmation of death were censored at the last date they were known to be alive. Time to response was defined as the time from the first dosing of GC1118 to the date of the first radiological objective response (CR or PR), and duration of response as the time from the first radiological objective response to the date of the radiological confirmation of PD. Tumor response was assessed by the investigators according to RECIST 1.1 every 6 weeks. Anti-drug antibody was measured from the serum samples collected at baseline and every 6 weeks thereafter. RAS/BRAF mutations and EGFR status results were verified at baseline using local test results, and if such data were unavailable, a molecular screening was performed using archival or newly obtained tumor specimens before administering the study treatment. For exploratory analysis of circulating tumor DNA (ctDNA) and EGFR-ligands, a 6 mL of plasma sample and a 5 mL of serum sample were collected respectively before the first dosing of the study drug. The ctDNA sequencing was carried out using Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific, Inc., Waltham, MA) to detect hot spot mutations of EGFR, KRAS, NRAS, HRAS, BRAF, PIK3CA, and PTEN. Sequencing data were analyzed with Torrent Suite ver. 5.0.4.
For pharmacokinetic (PK) analysis, serum samples were obtained at the following time points during the phase 1b study: 0, 0.5, 1, 2, 4, 8, 12, 24, 72, and 120 hours after the first dosing of GC1118 on the first and third cycles. In addition, pre-dosing samples were collected within seven hours before each dosing of GC1118 from all patients during both study phases.
Serum concentrations of GC1118 and its anti-drug antibody were determined at the Department of Clinical Pharmacology and Therapeutics, Seoul National University Hospital (Seoul, Korea), and ctDNA and EGFR-ligands were analyzed at Macrogen, Inc. (Seoul, Korea). Clinical laboratory tests and tissue biomarker analysis were performed at the local laboratory of each participating center.
4. Statistical analysis
Following the standard 3+3 design, a minimum of 3 or up to 12 evaluable patients in each dose group were planned to be enrolled in the phase 1b study. An evaluable patient was defined as an individual who had at least 2 weeks of follow-up data after completing a minimum of two cycles of treatment (four injections of GC1118 and two cycles of FOLFIRI) or experienced a DLT. For the phase 2a study of Simon’s optimal two-stage design, a total of 29 patients, including a minimum of nine patients in the first stage and 20 additional patients in the second stage, were required to test the true response rate at the significance level of 5% with 80% power, assuming 10.0% ORR (H0) versus 31.7% (H1). To proceed to the second stage, at least two responders out of nine patients in the first stage were required. GC1118 would be considered of potential clinical interest if there were six or more responders out of the total of 29 treated patients.
Patient demographics and baseline characteristics were summarized with descriptive statistics. Relative dose intensity (RDI) was calculated by dividing the actual cumulative dosage received by the planned dosage based on the number of doses the patient was scheduled to receive. Adverse events were coded using MedDRA ver. 24.0. The TEAE was classified as an adverse drug reaction (ADR) if the causal relationship was assessed as one of the following categories: definitely related, probably or likely related, possibly related, unlikely related, or unassessable. The severity of an adverse event was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events ver. 4.03. The tumor response rate from the phase 1b study was presented as a percentage with confidence interval (CI) using the exact method, whereas, for the data from the phase 2a study, a uniformly minimum variance unbiased estimator was calculated. Both 90% and 95% CIs were calculated; however, 95% CIs are presented in this article. Survival outcomes were analyzed using the Kaplan-Meier method, and restricted mean survival time (RMST) was also calculated. For an exploratory purpose, a hazard ratio (HR) for PFS in various subgroups was calculated using Cox proportional regression analysis. These subgroups were determined based on the sidedness of CRC, ctDNA mutation status, level of serum EGFR-ligand, and intensity of EGFR expression in tumor tissue. All statistical analyses were performed using the Statistical Analysis Software (SAS) ver. 9.4 (SAS Institute Inc., Cary, NC).
Based on serum PK concentration data, the following PK parameters were derived using a non-compartmental analysis method implemented in Phoenix WinNonlin ver. 6.4 (Certara, St. Louis, MO): maximum serum drug concentration (Cmax); maximum steady-state drug concentration (Cmax,ss); time to reach Cmax (tmax); time to reach maximum drug concentration at steady state (tmax,ss); drug concentration observed at the last planned timepoint prior to dosing (Ctrough); area under the serum concentration-time curve (AUC) from dosing to the time of the last measured concentration (AUC0-168h); AUC from zero to infinity (AUCinf); elimination half-life (t1/2); effective half-life (t1/2 eff); and mean residence time.
Results
1. Patients
The study was conducted in six hospitals in South Korea from April 2018 to August 2021. In the phase 1b part of the study, three patients were initially enrolled to receive 3 mg/kg of GC1118 in combination with FOLFIRI. As one patient experienced a DLT event, three more patients were enrolled in the same dose group, followed by one more patient to replace the patient who withdrew from the study before any treatment response evaluation. The DMC decided 3 mg/kg to be the RP2D of GC1118, and further dose escalation was not proceeded (Fig. 1). All seven patients were included in the analyses of safety, PK, and immunogenicity of GC1118. In the efficacy analysis, six patients were included after excluding the patient whose post-treatment tumor assessment was not performed.
In the phase 2a study, a total of 24 patients were enrolled (n=9 in stage 1, n=15 in stage 2) (Fig. 1). Three out of nine patients in stage 1 showed objective tumor response and the study proceeded to stage 2. The enrollment for stage 2 was prematurely stopped after a total of 24 patients were enrolled before reaching the initial target of 29 patients. This decision was made because the number of responders, specifically 10 out of 24 patients, surpassed the minimum threshold (6 out of 29 patients) required to reject the null hypothesis.
The patient characteristics in each phase of the study are presented in Table 1. Out of seven patients in the phase 1b study, one patient had breast cancer, one had biliary tract cancer, and the other five had CRC, all of which were left-sided. Whereas in the phase 2a study, 66.7% (16/24) had left-sided CRC (from splenic flexure to rectum), and 33.3% (8/24) had right-sided CRC (from ascending colon to transverse colon). No RAS or BRAF mutations were detected in the tumor tissues (Table 1).

Demographics and baseline characteristics of patients
2. Treatment exposure
In the phase 1b study, patients received a median of 10 treatment cycles both with GC1118 (range, 1 to 26) and FOLFIRI (range, 1 to 19). The RDIs of GC1118 and the three drugs of FOLFIRI were generally high, with the medians ranging from 71.99% to 79.17% (Table 2).
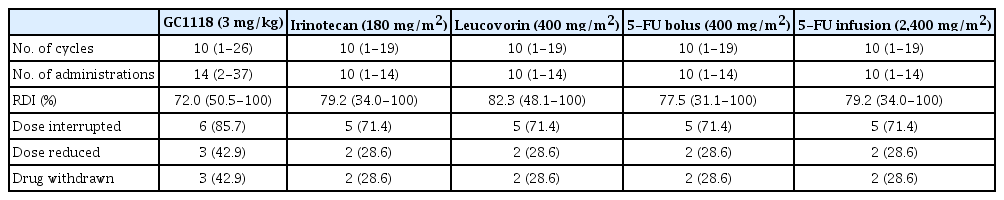
Exposure to treatment in phase 1b
Patients in the phase 2a study received slightly more cycles of treatment than those in the phase 1b study. The median number of GC1118 cycles administered was 13 (range, 2 to 45). The number of FOLFIRI cycles administered ranged from 2 to 42, with a median number of cycles of 12, except for irinotecan, for which it was 11.5. The median RDI for GC1118 was 65.7%, whereas the median RDI for the chemotherapeutics of the FOLFIRI regimen ranged from 69.4% to 75.7% (Table 3).

Exposure to treatment in phase 2a
3. Safety
Six patients (85.7%) in the phase 1b study experienced TEAEs and ADRs. There was one DLT, a grade 4 neutropenia that lasted longer than 7 days, in one of the first three patients enrolled. The dosing of GC1118 was temporarily withdrawn at least once in the six patients (Table 2), primarily due to neutropenia (4 patients) or rash (3 patients). Three patients were permanently discontinued from GC1118 treatment due to an event of maculo-papular rash, dry skin, or ileus, respectively. The most common ADRs (all grades) were neutropenia (71.4%), diarrhea (71.4%), stomatitis (42.9%), upper abdominal pain (42.9%), rash (42.9%), and dry skin (42.9%) (Table 4). ADRs of grade 3 or 4 in severity were reported in five patients (71.4%), with neutropenia (71.4%) being the most common event (Table 5).
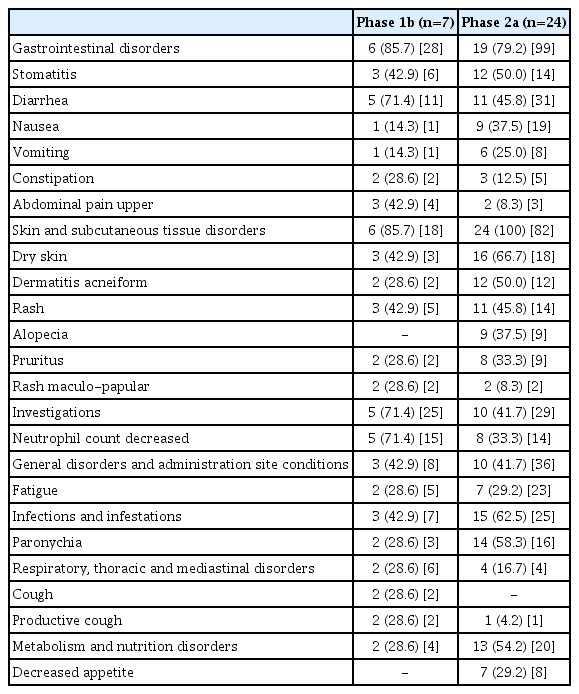
Adverse drug reactions occurring in ≥ 25% of either of the study population
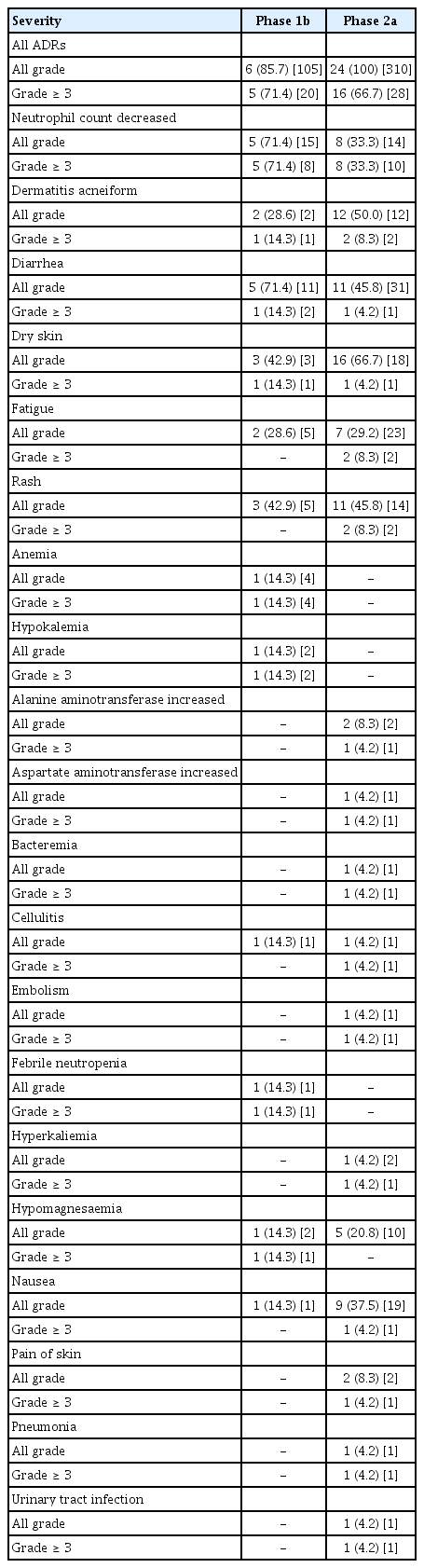
GC1118-related grade 3/4 adverse drug reactions
During the phase 2a study, all 24 patients (100%) experienced TEAEs and ADRs. In three patients (12.5%), a total of four serious adverse events were reported, including large intestinal obstruction, nausea, bacteremia, and pneumonia. Among these four serious events, bacteremia and pneumonia that occurred in one patient were classified as ADRs. The bacteremia was determined to be probably related to GC1118, whereas the causality of pneumonia to GC1118 was assessed to be unlikely related. In 18 patients (75%), at least one dosing of GC1118 was temporarily withdrawn (Table 3), and the common TEAEs that caused the dose interruption included neutropenia (8 patients), rash (8 patients), stomatitis (6 patients), fatigue (5 patients), and dry skin (5 patients). GC1118 was permanently discontinued in five patients (21%) due to the following TEAEs: dermatitis acneiform, fatigue, paronychia, and pneumonia in four patients each, and skin erosion and skin fissure in one patient.
Common ADRs (all grades) were dry skin (66.7%), paronychia (58.3%), dermatitis acneiform (50.0%), stomatitis (50.0%), rash (45.8%), diarrhea (45.8%), nausea (37.5%), alopecia (37.5%), pruritus (33.3%), and neutropenia (33.3%) (Table 4). When the ‘rash,’ ‘rash maculo-papular,’ and ‘dermatitis acneiform’ were grouped into the single term, acneiform rash, the most frequent ADR was identified as acneiform rash, occurring in 95.8% of all patients.
ADRs of grades 3 or 4 were reported in 16 patients (66.7%), with neutropenia (33.3%) being the most common event (Table 5). No abnormal finding in the electrocardiogram was found, and there was no treatment-related death during either of the study phases.
4. Immunogenicity
None of the patients in both phases of the study had a positive result for anti-drug antibody in the blood samples collected either during the pre-treatment or post-treatment period.
5. Efficacy outcomes
Out of six patients with evaluable efficacy assessment data in the phase 1b study, none achieved CR, and one with CRC had PR, as a result, the ORR was 16.7% (95% CI, 0.4 to 64.1). Meanwhile, four patients had stable disease, and the DCR was 83.3% (95% CI, 35.9 to 99.6) (Table 6, Fig. 2A).

Efficacy results
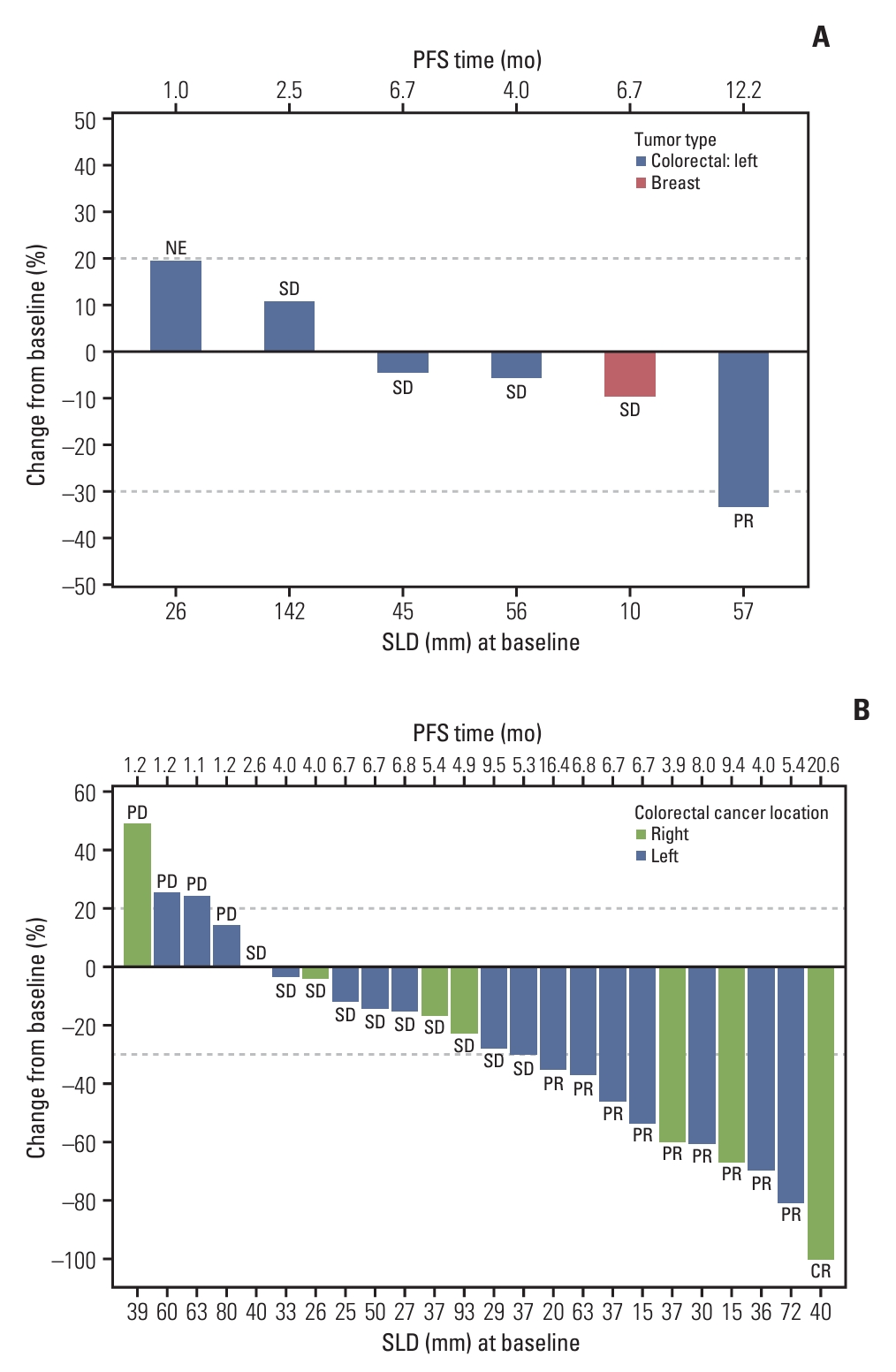
Waterfall plots showing individual patient’s maximum percent change in tumor size from baseline in phase 1b (A) and phase 2a (B). CR, complete response; NE, not evaluable; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease; SLD, the sum of the longest diameter of target lesions. Each dashed line above and below 0 indicates a 20% increase and a 30% decrease in SLD from baseline, respectively.
In the phase 2a study, the ORR was 42.5% (95% CI, 23.5 to 62.0), and the DCR was 83.6% (95% CI, 51.8 to 97.2) (Table 6, Fig. 2B). The median PFS and OS were 6.7 months (95% CI, 4.0 to 8.0) and 25.4 months (95% CI, 14.3 to not reached), respectively (Fig. 3). The medians of time to response and duration of response were 5.3 months (range, 1.1 to 9.5 months; 95% CI, 1.3 to not reached) and 6.8 months (range, 1.4 to 16.7 months; 95% CI, 2.8 to 13.7), respectively (Table 6). Survival outcomes based on the RMST method from each phase of the study are also provided in Table 6.

Kaplan-Meier curves of progression-free survival (A) and overall survival (B) from phase 2a. CI, confidence interval; NR, not reached.
6. Exploratory outcomes
Due to an insufficient number of patients in the phase 1b study for the planned exploratory subgroup analysis, subsequent analyses for exploratory outcomes were conducted only using the data from the phase 2a study. Among 16 patents with left-sided CRC, the ORR was 43.8% (PR=7). Meanwhile, the ORR was 37.5% (CR=1, PR=2) in the right-sided CRC (n=8). Cox regression analysis showed that the sidedness of CRC did not affect the PFS outcome: HR in the right-sided group to left-sided group was 1.0 (95% CI, 0.4 to 2.8).
Despite the absence of RAS or BRAF mutations detected from the tumor tissues, exploratory analysis of plasma ctDNA indicated that four patients (16.7%) in the phase 2a study harbored mutations in the downstream signaling pathways of EGFR, which included NRAS (p.Gln61Leu and p.Gln25Arg), PIK3CA (p.Glu545Lys, p.Arg88Gln, and p.His1047Arg), and PTEN (p.Leu171fs) mutations. However, the ctDNA mutation status was not associated with shorter PFS. No significant difference in PFS was observed according to the level of EGFR-ligands expression, such as amphiregulin or epiregulin, or the intensity of EGFR expression in tumor tissue (S4 Fig.). As for the distribution of the best overall responses according to the grade of skin toxicity, no clear trend could be deduced as the skin toxicity was centered in grade 2 and the number of patients was not large enough for proper interpretation of the data (S5 Fig.).
7. Pharmacokinetics
After a single intravenous injection of GC1118, Cmax (46.5 μg/mL) was reached at a median of 1.5 hours. The serum concentration-time curve showed a biphasic elimination pattern, with an elimination half-life of 81.8 hours (Table 7, S6 Fig.). At the steady state, the median Tmax was 1.5 hours, the mean Cmax was 69.7 μg/mL, and the effective half-life was 109.2 hours (Table 7, S6 Fig.). The trough concentration of GC1118 steadily increased until day 21 (in phase 2a) or day 28 (in phase 1b) and reached a steady state thereafter, with a mean trough level of 23.2 μg/mL (Table 7, S7 Fig.).
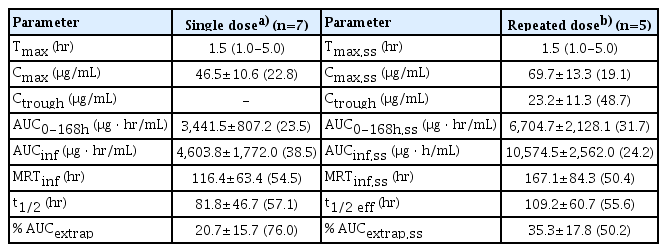
Pharmacokinetic parameters of GC1118 from phase 1b study
Discussion
In this open-label prospective study, GC1118 (3 mg/kg), a novel anti-EGFR monoclonal antibody, was administered weekly in combination with FOLFIRI to Korean patients with recurrent or metastatic solid tumors (phase 1b) and CRC (phase 2a). GC1118 was shown to have a similar efficacy as other currently available monoclonal antibodies against EGFR, and its safety profile was within a spectrum of the drug class.
In a previous phase 1 study involving patients with refractory solid tumors, the MTD for GC1118 monotherapy was determined at 4 mg/kg, with DLT appearing at 5 mg/kg [14]. However, taking into account the gradual increase in skin toxicity observed with continuous treatment at 4 mg/kg in the above-mentioned study [14], the RP2D of GC1118 for the present phase 2a study was eventually chosen at 3 mg/kg.
The ORRs in the present study were 16.7% and 42.5% for phase 1b and phase 2a, respectively, with the DCRs of 83.3% and 83.6%, respectively. The median PFS and OS in the phase 2a part were 6.7 months and 25.4 months, respectively. The efficacy outcomes reported above for phase 2a are evidently better than the results obtained in a cohort of anti-EGFR treatment-naïve CRC patients included in the cohort expansion part of a previous phase 1 study of GC1118 monotherapy, in which GC1118 (4 mg/kg) was used in patients with metastatic CRC who received no prior EGFR antibody treatment and who failed on 5-FU, oxaliplatin, and irinotecan treatment; the ORR for that cohort was 0%, with the DCR of 58.3% and median PFS of 14 weeks [14]. The efficacy outcomes of the present study are similar to previous trials using the combination of FOLFIRI with panitumumab as second-line therapy in metastatic CRC (ORR, 32-35%; median PFS, 5.9 to 7.7 months) [16,17].
Overall, GC1118 was well tolerated by the study patients. No treatment-related deaths were recorded. The most common treatment-related adverse event (AE) was skin toxicity, found in all patients included in the phase 2a study. Gastrointestinal AEs, stomatitis and diarrhea, were found in 50% and 45.8% of the patients, respectively. This safety profile differs slightly from the data from the GC1118 monotherapy phase 1 study, where the occurrence of stomatitis and diarrhea was observed in 21% and 8% of the patients, respectively [14]. Perhaps, the higher incidence of gastrointestinal AEs in the present study could result from the concomitant administration of FOLFIRI.
Importantly, none of the study patients developed antidrug antibodies at the end of the treatment phase. Unlike other available anti-EGFR antibodies, GC1118 exerts a potent inhibitory effect not only on low- but also on high-affinity ligand-induced EGFR signaling and proliferation [9]. Meanwhile, the high-affinity ligands, such as transforming growth factor alpha and heparin-binding epidermal growth factor-like growth factor, were implicated to play a role in acquired resistance to cetuximab and panitumumab [8]. This opens a perspective of administering GC1118 as another line of therapy in patients with innate or acquired resistance to other anti-EGFR antibodies [12,14].
Previous studies showed that GC118 has a non-linear PK profile, with systemic exposure increasing in a greater-than-dose-proportional manner [14]. Nevertheless, the present study demonstrated that GC1118 produced a potent antitumor effect even after 3 mg/kg, similar to that obtained with other anti-EGFR antibodies [16,17]. Furthermore, in a recent PK study involving patients with solid tumors, the mean clearance of GC1118 remained stable beyond the dose of 3 mg/kg. This implies that EGFR was fully saturated by GC1118 at ≥ 3 mg/kg, meaning that the inhibitory effect of the drug is unlikely to increase with further escalation of the dose [18].
Obviously, the results presented herein were obtained in a small group of patients, and as such, should be interpreted with caution until verified in a phase 3 trial. Nonetheless, our findings suggest that GC1118 administered weekly at 3 mg/kg in combination with FOLFIRI appears to be an effective and safe option in recurrent or metastatic CRC and merits further evaluation in this setting.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
The study protocol was reviewed and approved by the institutional review board at each institution (Asan Medical Center: 2018-1553, Chonnam National University Hwasun Hospital: CNUHH-2019-216, National Cancer Center: NCC2019-0286, Seoul National University Hospital: 1712-102-908, Seoul National University Bundang Hospital: B-1803-454-403, Yonsei University Severance Hospital: 4-2018-1139). All patients enrolled in this study provided informed consent to participate in this trial. This study was performed in accordance with the Declaration of Helsinki.
Author Contributions
Conceived and designed the analysis: Bang YJ.
Collected the data: Lee KW, Han SW, Kim TW, Ahn JB, Baek JY, Cho SH, Lee H, Kim JW (Jin Won Kim), Kim JW (Ji-Won Kim), Kim TY, Hong YS, Beom SH, Cha Y, Bang YJ.
Contributed data or analysis tools: Lee KW, Han SW, Kim TW, Ahn JB, Baek JY, Cho SH, Lee H, Kim JW (Jin Won Kim), Kim JW (Ji-Won Kim), Kim TY, Hong YS, Beom SH, Cha Y, Bang YJ.
Performed the analysis: Lee H, Kim S, Lee KW, Han SW, Choi Y.
Wrote the paper: Lee KW, Han SW, Choi Y, Lee H.
Writing - review and editing: Lee KW, Han SW, Kim TW, Ahn JB, Baek JY, Cho SH, Lee H, Kim JW (Jin Won Kim), Kim JW (Ji-Won Kim), Kim TY, Hong YS, Beom SH, Cha Y, Bang YJ.
Conflicts of Interest
Keun-Wook Lee reported research funding (to his institution for conducting clinical trials) from GC biopharma, AstraZeneca, Ono Pharmaceutical, Merck, Sharp, and Dohme, Merck KGaA, Pfizer, BeiGene, Zymeworks, ALX Oncology, Astellas, Macrogenics, Amgen, Seagen, Bolt Therapeutics, Trishula Therapeutics, Daiichi Sankyo, Taiho Pharmaceutical, InventisBio, Leap Therapeutics, MedPacto, LSK BioPharma, Y-BIOLOGICS, Metafines; consulting fees from Metafines, Astellas, Daiichi Sankyo, Ono pharmaceutical, Sanofi-Aventis, Guardant Health (outside the submitted work); and honoraria from Ono pharmaceutical, Boryung, Daiichi Sankyo, Astellas, Sanofi-Aventis (outside the submitted work). Sae-Won Han has received honoraria from IMBdx, Ono Pharmaceutical; and research funding (institution) from IMBdx, Hanmi, Loxo, Roche, Genentech, Mirati Therapeutics, Turnstone Bio, Arcus Biosciences, MSD, BeyondBio, Jeil Pharmaceutical Co, Janssen, Seagen, Lilly, MedImmune, Leap Therapeutics. Yung-Jue Bang has received compensation for consulting services or participation in advisory boards from Astellas, Amgen, Samyang Biopharm, Hanmi, Daewoong, SK Biopharm. This study was funded by GC Biopharma Corp., Yongin, South Korea.
Acknowledgements
We thank the patients and their families, all sub-investigators, and study teams at each of the participating centers.