Genomic Landscape of Pulmonary Sarcomatoid Carcinoma
Article information

Abstract
Purpose
Pulmonary sarcomatoid carcinoma (PSC) is a rare aggressive subtype of non–small cell lung cancer (NSCLC) with limited therapeutic strategies. We attempted to elucidate the evolutionary trajectories of PSC using multiregional and longitudinal tumor samples.
Materials and Methods
A total of 31 patients were enrolled in this study and 11 longitudinal samples were available from them. Using whole exome sequencing data, we analyzed the mutational signatures in both carcinomatous and sarcomatous areas in primary tumors of the 31 patients and longitudinal samples obtained from 11 patients. Furthermore, digital droplet polymerase chain reaction (ddPCR), and programmed death-ligand 1 (PD-L1) immunohistochemistry using the Ventana SP263 assay were performed.
Results
TP53 was identified as the most frequently altered gene in the primary (74%) and metastatic (73%) samples. MET exon 14 skipping mutations, confirmed by ddPCR, and TP53 mutations were mutually exclusive; whereas, MET exon 14 skipping mutations frequently co-occurred with MDM2 amplification. Metastatic tumors showed dissimilar genetic profiles from either primary component. During metastasis, the signatures of APOBEC decreased in metastatic lesions compared with that in primary lesions. PSC showed higher MET and KEAP1 mutations and stronger PD-L1 protein expression compared with that recorded in other NSCLCs.
Conclusion
Decreased APOBEC signatures and subclonal diversity were detected during malignant progression in PSC. Frequent MET mutations and strong PD-L1 expression distinguished PSC from other NSCLCs. The aggressiveness and therapeutic difficulties of PSC were possibly attributable to profound intratumoral and intertumoral genetic diversity. Next-generation sequencing could suggest the appropriate treatment strategy for PSC.
Introduction
Pulmonary sarcomatoid carcinoma (PSC), a subtype of non–small cell lung cancer (NSCLC), is characterized by a biphasic phenotype comprising coexisting carcinomatous and sarcomatous components. This rare entity, showing aggressive behavior, accounts for only 0.1%-0.4% of all lung cancers [1]. Patients diagnosed with PSC have a poor prognosis associated with a high rate of progression and an average survival of 8-16.4 months [2,3]. Currently, PSC is generally treated following guidelines for NSCLC treatments. Although surgery is the best treatment option, most patients are diagnosed at an advanced stage of the disease, and the survival benefit of conventional chemotherapy regimens remains controversial [4].
According to the 2021 World Health Organization classification, PSC can be histologically divided into five subtypes [1]. Pleomorphic carcinoma, the most prevalent subtype, is defined as an NSCLC that contains at least 10% of morphologically distinct mesenchymal (spindle and/or giant) cancer cells. If these spindle or giant cells comprise the entire tumor, they are referred to as spindle cell carcinomas or giant cell carcinomas, respectively. Carcinosarcomas contain heterologous elements, such as rhabdomyosarcoma, chondrosarcoma, and osteosarcoma, mixed with NSCLC. The pulmonary blastoma consists of fetal adenocarcinoma and primitive mesenchymal stroma.
The biphasic nature of PSC is currently considered a result of epithelial-mesenchymal transition (EMT), which is defined as the complex cellular event resulting in the transformation of an epithelial cell to a mesenchymal cell characterized by altered morphology, the gain of motility, loss of cell polarity, and immunohistochemical markers such as vimentin expression or E-cadherin loss. Transcription factors, mainly SNAIL, TWIST, and ZEB, play important roles in the activation of EMT. In cancer cells, mutations in any one of these transcription factors can lead to epithelial-mesenchymal plasticity, inducing an intermediate phenotype that can effectively induce cancer cells to invade, metastasize, and develop chemoresistance [5].
Recently, research on PSC has revealed interesting findings. Next-generation sequencing has revealed mutations in many genes, including MET exon 14 skipping mutations, in PSC [2,6-9]. In addition, PSC frequently (25%-90.2%) express programmed death-ligand 1 (PD-L1) at a high level, and a few studies have claimed that high PD-L1 expression is associated with tumor recurrence or metastatic lymph nodal status [2,4,10]. Moreover, patients with PSC, successfully treated with anti–programmed death-1/PD-L1 antibodies, were previously reported [11]. These findings may lead to new prospects and insights into therapeutic strategies for PSC.
Therefore, we aimed to explore the genomic landscape and evolutionary trajectories of PSC by whole exome sequencing (WES) of PSC using multiregional and longitudinal tumor samples. Furthermore, we attempted to gain insight into the morphological heterogeneity of tumor plasticity and the evolution of PSC.
Materials and Methods
1. Design of study cohort
A total of 47 patients diagnosed with PSC via surgical resection at Seoul National University Bundang Hospital (SNUBH), Seongnam, Korea, between 2006 and 2018 were selected for this study. Patients, with a previous history of malignancy, a follow-up period shorter than two years, or who received preoperative treatment, were excluded. Moreover, those with an insufficient amount of tissue available for molecular testing were excluded. Finally, 31 patients with primary PSC were enrolled in this study. Longitudinal samples of 11 patients were available, which showed regional lymph node metastasis (n=10) or distant metastasis (n=1) (Fig. 1). Clinicopathological information was obtained through retrospective review of medical records. This study was conducted following the Declaration of Helsinki and the Guidelines for Good Clinical Practice. Hematoxylin and eosin (H&E)–stained slides of the resected specimens were reviewed by two pathologists (H.J. Kwon and J.H. Chung). Pathological diagnosis and histological subtypes were identified based on the 2021 World Health Organization Classification of Lung Tumors [1]. Based on the 8th edition of the American Joint Committee on Cancer staging manual, the pathological stages were detected [12].
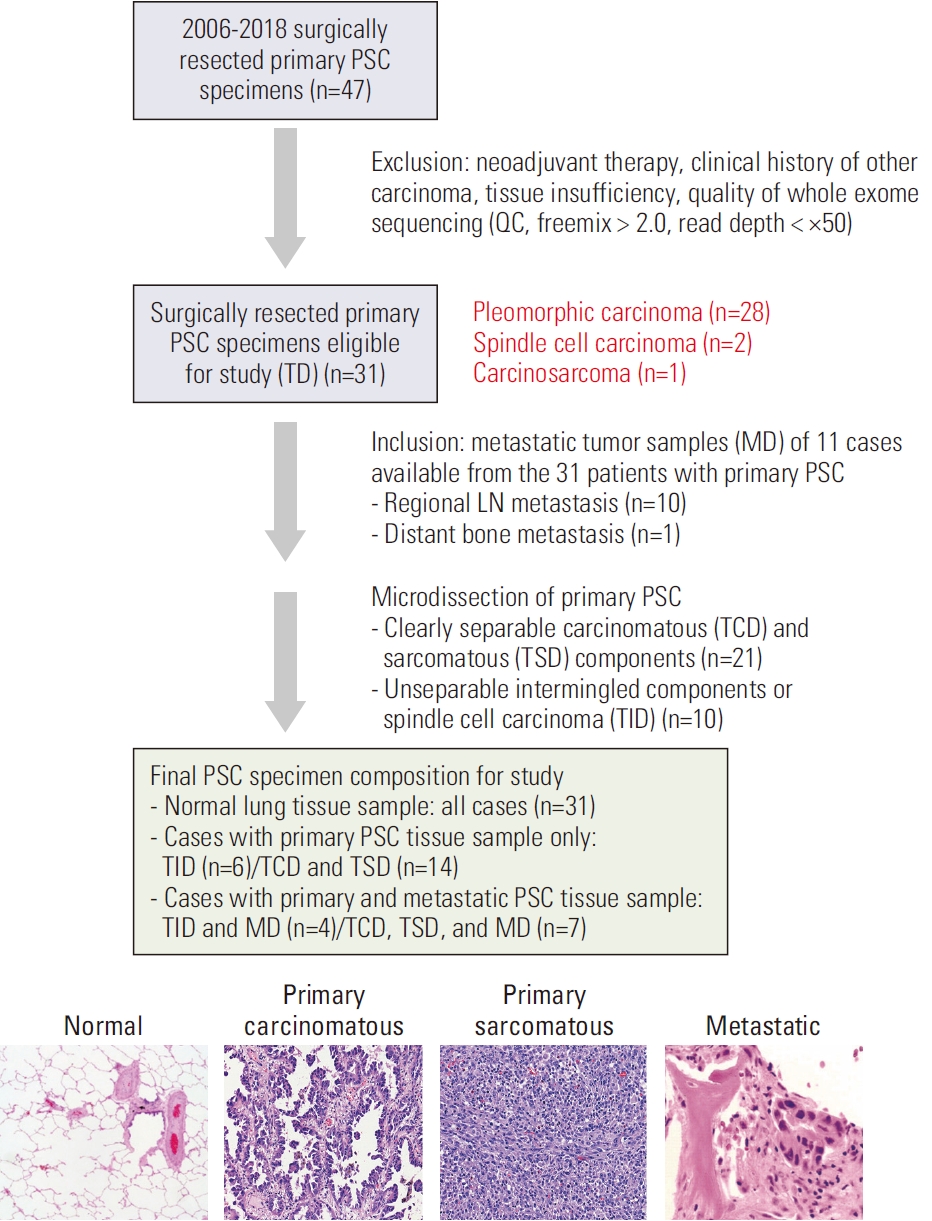
Study design. LN, lymph node; PSC, pulmonary sarcomatoid carcinoma; QC, quality control; TCD, carcinomatous component of the primary tumor; TD, primary tumor; TID, primary tumor with inseparable components; TSD, sarcomatous component of the primary tumor.
2. WES of PSC using formalin-fixed paraffin-embedded tumor samples
A total of 63 samples were analyzed, which included 31 primary PSC samples (hereafter referred to as ‘TD’, comprising 21 pairs of samples from carcinomatous and sarcomatous areas of primary biphasic PSC, 2 samples of spindle cell carcinomas and 8 samples of biphasic PSCs with inseparable components), and metastatic lesions (hereafter referred to as ‘MD’) from 11 patients. Both carcinomatous areas (hereafter referred to as ‘TCD’) and sarcomatous areas (hereafter referred to as ‘TSD’) of the primary tumors were carefully marked on H&E slides and micro-dissected accordingly. Normal lung tissue blocks were included in each case.
DNA was extracted from the tumor samples, and libraries were constructed using the Agilent SureSelect V6-Post (formalin-fixed paraffin-embed [FFPE]) Target Enrichment Kit (Santa Clara, CA). The constructed libraries were amplified and sequenced using an Illumina platform sequencer (Illumina, San Diego, CA), then aligned to the hg19 genome reference using MEM algorithm in BWA 0.7.12. The generated SAM/BAM files were sorted and marked for duplications using SAMTOOLS v1.9 and Picard 1.13, respectively. The IndelRealigner from GATK 3.4.0 was used for correcting small insertion/deletions (Indels) based on known germline variations from the gatk bundle release. The mean depth of coverage was ×128.
3. Analyzing WES data for genetic alterations
Single nucleotide variation (SNV) and Indels were detected using Mutect2 from GATK4 v4.0.8, and annotated using Annovar. Falsely detected variants were filtered out using FilterMutectCalls from GATK4. Synonymous mutations and mutations in intron or intergenic regions were also filtered out. The detected genetic alterations that met all the following criteria were further analyzed: (1) read depth more than ×20, (2) variable allelic frequency (VAF) higher than 0.05, and (3) allelic depth more than ×5. EMT-related genes were further selected [13,14] and analyzed using ConsensusPathDB (http://cpdb.molgen.mpg.de/).
Copy number variation (CNV) was analyzed using the following recommended workflow. Firstly, gc_wiggle and bam2seqz function from sequenza-utils were applied on both tumor and normal bam files to generate seqz files, and binning seqz file into small regions. Then, the sequenza.extract function in R sequenza package [15] was used to normalize the sequencing depth and to segment copy number region. The cellularity and ploidy information was also obtained using the sequenza.fit function. Finally, GISTIC2 was applied to detect the recurrently altered copy number regions.
4. Droplet digital polymerase chain reaction for MET exon 14 skipping mutation
RNA was extracted from FFPE tumor tissues using a Maxwell CSC RNA FFPE Kit (Promega Inc., Madison, WI) and quantified using a Nanodrop (Thermo Fisher Scientific, Waltham, MA) according to the protocol provided by the manufacturer (Gencruix Inc., Seoul, Korea), then stored at –80°C in a deep freezer. The Droplex cMET Exon14 Skipping Kit was used to perform a two-step process: reverse transcription and digital PCR.
QuantaSoft software (Bio-Rad, Hercules, CA) was manually used to read droplet digital polymerase chain reaction (ddPCR). The FAM channels of OM1 and OM2 represent the copy number of the cMET exon14 skipping mutation and total cMET, respectively, which were used to calculate the mutation index (MI%). Greater details of ddPCR are provided in the Supplementary Methods section.
5. Hot spot mutation testing for EGFR and KRAS
To detect epidermal growth factor receptor (EGFR) mutations in exons 18-21 and KRAS mutations in codons 12, 13, and 61, EGFR and KRAS were analyzed at the time of diagnosis using a PCR-based assay (PANAMutyper, Panagene, Daejeon, Korea) according to the protocol provided by the manufacturer.
6. Phylogenic analysis
Phylogenic analysis was performed considering nonsynonymous mutations in seven PSC cases that had three types of tumor samples: TCD, TSD, and MD. The results are depicted as a phylogenic tree composed of a trunk and branches representing overlapping mutations found in all three types of tumors and those either common to primary PSC samples or specific to an individual sample, respectively.
7. PD-L1 immunohistochemistry
For PD-L1 immunohistochemistry, a Ventana SP263 assay (Ventana Medical Systems, Inc., Tucson, AZ) was used. Briefly, the slides were stained with anti–PD-L1 SP263 rabbit monoclonal primary antibodies using the Benchmark ULTRA system, following the manufacturer’s instructions; negative control agents were included in the analysis for the comparative study. Three parameters were recorded for PDL1 in each case, such as the total tumor area, carcinomatous area, and sarcomatous area. The percentage of the number of stained tumor cells in the overall tumor area (tumor proportion score [TPS]) was determined and positive expression was defined as TPS higher than 50.
8. Statistical analysis
Statistical analyses were performed using SPSS Statistics ver. 25.0 (IBM Corp., Armonk, NY) and the R program (R Foundation for Statistical Computing, Vienna, Austria). Pearson’s correlation and linear regression were used to evaluate the association between PD-L1 expression and histologic components of PSC. Survival analysis was performed using the log-rank test. For all analyses, statistical significance was considered at p < 0.05.
Results
1. Clinicopathologic characteristics of the study cohort
The clinicopathological characteristics of the cohort are summarized in S1 Table. The patients, with an average age of 68 years, included 80.7% of men with an active smoking history (92.0% of the male patients). Pleomorphic carcinoma was found to be the dominant subtype (28/31, 90.3%), followed by less abundant spindle cell carcinoma (2/31, 6.5%) and carcinosarcoma (1/31, 3.2%) (S2 Fig.). PSC frequently invaded the pleura (58.1%), lymphatics, or blood vessels (67.7%), reflecting its aggressive behavior. Mostly, the patients were at pathological stage 2 or 3 during surgery. A subset of tumors was tested for EGFR (2/17, 11.8%) and KRAS (2/11, 18.2%) mutations using pyrosequencing according to the physician’s request at the time of diagnosis.
Patients were followed-up for 1-176 months (median of recorded follow-up time: 38 months) after surgery. Approximately half of the 31 patients (51.6%) diagnosed with PSC eventually developed metastasis to distant sites excluding regional lymph nodes, such as the bone, adrenal gland, and brain. The median time required for disease progression was 9 months. Seventeen patients (54.8%) died within their follow-up period; 12 patients (38.7%) from disease progression and five patients from surgical complications or other causes. The 1-year and 5-year overall survival (OS) rates of the patients with PSC were 64.5% and 48.2%, respectively, with a median OS of 24 months. Twenty-one patients received adjuvant chemotherapy, which frequently included cisplatin and vinorelbine (NP), as first-line treatment. Two patients were enrolled in immunotherapy clinical trials immediately after surgery, whereas, eight patients did not receive additional adjuvant treatment due to informed refusal or underlying chronic obstructive pulmonary disease.
2. Mutational landscape of primary PSC detected by WES
WES revealed heterogeneous molecular characteristics of primary (TD samples) (Fig. 2A) and metastatic PSC (MD samples) (Fig. 2B). To compare each primary PSC with one another, the genetic characteristics of each individual primary PSC were examined within the entirety of the primary tumor itself, as illustrated in Fig. 2A, rather than its components. TP53 was the most frequently altered gene in both the primary (77%) and metastatic (73%) PSC and all of its mutations were non-recurrent and representing a unique mutation site in each tumor (Fig. 2C). TP53 mutations included missense mutations (13/31, 41.9%) that was detected frequently, deletions (8/31, 25.8%), and nonsense mutations (4/31, 12.9%).

Mutational landscape of pulmonary sarcomatoid carcinoma. Whole exome sequencing results of primary tumors (A) and metastatic tumors (B). Lollipop plot of the most commonly mutated gene, TP53, in both primary and metastatic PSC (C). MET exon 14 skipping mutations, validated by digital droplet polymerase chain reaction, were present in five cases (D). Distant metastasis, metastasis to sites other than regional lymph nodes, detected clinically; MD, metastatic tumor; PD-L1, programmed death-ligand 1; PSC, pulmonary sarcomatoid carcinoma; TD, primary tumor including all histologic components; TMB, tumor mutation burden; TPS, tumor proportion score.
In primary PSC, 12 cases (38.7%) harbored pathogenic NSCLC driver mutations in genes such as EGFR (5/31, 16.1%), KRAS (2/31, 6.5%), and MET (5/31, 16.1%) (S3 Table). These cases were mostly pleomorphic carcinoma, with adenocarcinoma as their carcinomatous components. Using pyrosequencing performed at the time of diagnosis, the same EGFR and KRAS mutations were confirmed; moreover, it confirmed the KRAS mutation in the metastatic counterpart of a tumor with a low VAF (3%) in WES.
MET alteration, including mutations and amplification, was detected in six primary PSC (19%) and in a metastatic sample (9%) (Fig. 2A and D). The seven tumor samples harboring MET mutation were subjected to ddPCR, which identified all as MET exon 14 skipping mutations (S3 and S4 Tables). These MET mutations included a novel deletion mutation, c.2901_2911del (p.E967fs), and a splice site mutation reported previously [16,17]. In this study, MET exon 14 skipping mutations and MET amplification co-occurred with TP53 in only one of the six primary PSC and not with any EGFR or KRAS mutations. However, it frequently co-occurred with MDM2 amplification (5/6, 83.3%) (Fig. 2A).
3. Evolutionary trajectories and mutational signatures during regional and distant metastasis
The mutational profiles of primary and metastatic PSCs showed little difference in the composition of the altered genes. However, recurrent amplification of MDM2 (19%) and EGFR (6%) was observed in the primary PSC rather than the metastatic samples. Tumor mutation burden (TMB) was significantly higher in primary tumors than in metastatic ones (median, 3.08/MB vs. 1.05/MB; p=0.006) and tended to be higher in current smokers than in never-smokers and ex-smokers (6.85/Mb vs. 2.29/Mb and 3.69/Mb, respectively), however, these smoking habit-associated differences were not statistically significant (p=0.363).
Comparative analyses of EMT signatures [13,14] revealed that TCD showed more frequent alterations in EMT-related genes, which were enriched in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, such as the phosphoinositide 3-kinase (PI3K)–AKT signaling pathway and development-associated gene ontology, compared with that in TSD. In contrast, TSD showed frequent alterations in the NOTCH signaling pathways of EMT-related genes. Metastatic samples showed frequent pathways similar to those of the TCD (S5 Table).
4. Molecular characteristics and phylogeny of micro-dissected PSC tumor components
Analysis on the molecular characteristics of primary biphasic PSCs, which could be micro-dissected according to histology, were performed in greater depth. TP53, CDH23, LRP1, and EGFR were the most frequently altered genes in both TCD and TSD, displaying a similar mutational landscape. Comparison of the biphasic components of primary PSC using GISTIC revealed that TSD had more amplification peaks than that of TCD (Fig. 3A and B). Both components showed amplification in the 7p11.2 and 12q15, where EGFR and MDM2 locate, respectively, and deletion in the 11p15.5 and 17q21.31, where HRAS and KANSL1 locate, respectively. In addition to these copy number alterations, TSD showed another amplification peak at 1q21.3.

Copy number variation analysis of micro-dissected carcinomatous and sarcomatous components in pulmonary sarcomatoid carcinoma. Copy number variation of primary carcinomatous (A) and primary sarcomatous (B) components. TCD, carcinomatous component of primary tumor; TSD, sarcomatous component of primary tumor.
Phylogenic study using seven cases consisting of three types of tissue samples (TCD, TSD, and MD) was performed. Metastatic samples in these cases originated from regional lymph node metastases, except for one bone metastasis (case SC13). All seven cases were pleomorphic carcinomas with a history of smoking. The TNM stage of these patients was 2 or 3 at the time of surgery.
Common and unique mutations in different components were compared (Fig. 4A). The two primary tumor components and the metastatic tumors had several unique as well as common mutations. Among these three components, TCD and TSD most frequently exhibited mutually shared common mutations.

Mutational profiling and phylogenic analysis of the selected seven pulmonary sarcomatoid carcinoma cases. Comparison of frequently mutated genes in the micro-dissected three tissue samples of each case (A). Phylogenic trees of representative cases, including case SC13 with no driver mutations, case SC14 with EGFR and TP53 mutation, and case SC33 with KRAS and TP53 mutation (B). Overlapping mutations in the three types of tumor samples are indicated by black trunks, and mutations common only to primary samples are indicated by red trunks. Mutations specific to TCD, TSD, and MD samples are indicated by orange, blue, and black lines, respectively. Length is proportional to the number of variants. ADC, adenocarcinoma; ASC, adenosquamous carcinoma; EGFR, epidermal growth factor receptor; MD, metastatic tumor; n, number of mutations; PD-L1, programmed death-ligand 1; SqCC, squamous cell carcinoma; TCD, carcinomatous component of primary tumor; TPS, tumor proportion score; TSD, sarcomatous component of primary tumor.
The phylogenetic trees of three representative cases are shown in Fig. 4B. The NSCLC driver mutations, such as EGFR or KRAS, were absent in the majority of the analyzed cases (5/7, 71.4%) as represented by case SC13. The three tissue samples of case SC13 showed several common genetic mutations, such as those in EPHA2, DNMT3A, and MUC5B, presented as a long common trunk.
Two patients had a pathogenic NSCLC driver mutation, EGFR and KRAS, respectively. These cases were characterized by a short common trunk that included a driver and concurrent TP53 mutations, suggesting that driver mutations contribute to early carcinogenesis. The metastatic sample of the EGFR-mutated case (case SC14) also harbored a TERT mutation that was absent in the primary tumor samples.
5. The COSMIC mutational signature of micro-dissected PSC tumor components
The COSMIC mutational signatures of the seven microdissected PSC cases were explored (Fig. 5). Signature 1, the most common recurrent signature, was found in the spontaneous deamination of 5-methycytosine; followed by the less frequent signatures 4 and 6, which were related to smoking and defective DNA mismatch repair, respectively. Unique signatures of individual cases were also detected, which included signature 16 that is common in liver cancer, and signature 18 that is frequent in neuroblastoma.

Mutational signature of the selected seven pulmonary sarcomatoid carcinoma cases. The APOBEC mutational signature (Signature 2) was characteristically absent or reduced in metastatic tumor samples. MD, metastatic tumor; TCD, carcinomatous component of primary tumor; TSD, sarcomatous component of primary tumor.
The comparative analyses of metastatic and primary tumors in each case revealed that the metastatic tumors universally exhibited lost or reduced relative contribution of signature 2 (p=0.016) (S6 Fig.). Signature 2, also known as the APOBEC signature, represents the activity of the AID/APOBEC family of cytidine deaminases characterized by C > T alterations.
6. Comparative analyses of frequently mutated genes in untreated primary NSCLC using WES
For further understanding the uniqueness of the mutational characteristics of PSC, we compared the WES results of primary PSC (TD samples) in this study with those of primary lung adenocarcinoma (LUAD) or lung squamous cell carcinoma (LUSC) obtained from The Cancer Genome Atlas (TCGA) data (http://www.cbioportal.org) and previous publications from Asia [18,19]. For this purpose, SNVs and Indels were considered. Fig. 6 displays the 11 most frequently mutated genes found in LUAD, LUSC, and PSC.

Comparative analysis of frequent mutations in untreated primary lung cancer using whole exome sequencing. The Cancer Genome Atlas (TCGA) data from cbioportal.org and East Asian data from two large cohorts [18,19] were compared to pulmonary sarcomatoid carcinoma whole exome sequencing results obtained in this study. SNV/INDEL were considered. INDEL, insertion and deletion; LUAD, lung adenocarcinoma; LUSC, squamous cell carcinoma; PSC, pulmonary sarcomatoid carcinoma; SNV, single nucleotide variation; VAF, variable allelic frequency.
In this study, PSC was distinguished from other NSCLCs by the highest MET mutation rate regardless of ethnicity. The mutation frequency of EGFR and KRAS in PSCs showed a similar tendency in Asian LUAD cases. However, the overall mutation profile was not identical to that of LUAD or LUSC cases with Asian or Western ethnicity.
7. PD-L1 immunohistochemistry in PSC
PSC expressed a high level of PD-L1 protein in both primary (TD samples, median TPS 85, PD-L1 positive in 61.3%) and metastatic tumors (median TPS 70, PD-L1 positive in 54.5%) (S7 Fig.). TSD showed stronger PD-L1 expression than that in TCD; however, the difference was not statistically significant (median TPS, 60 vs. 90; p=0.0928). In 11 cases with available metastatic tissue, MD expressed PD-L1 at an intermediate level between that evaluated in the primary tumor components.
In pleomorphic carcinoma, high PD-L1 expression (stronger than TPS 50) was significantly associated with the histological subtype of the carcinomatous component (p=0.029). Interestingly, the histological subtype of TCD was more significantly associated with PD-L1 positive expression of TSD than that of TCD itself (p=0.008 and p=0.061, respectively).
8. Molecular characteristics associated with tumor progression and patient survival
The association of the molecular characteristics of primary PSC to tumor progression and patient survival was also explored. Patients with PSC, who harbored a MET mutation, showed shorter median progression-free survival (PFS) (5 months vs. 35 months) and median OS (19 months vs. 39 months) than those without such mutation, however, this was not a statistically significant prognostic factor (p=0.193, PFS; p=0.621, OS). PSC that clinically developed distant metastasis had less frequent TP53 mutations compared to patients without distant metastasis (9/16, 56.3% vs. 14/15, 93.3%; p=0.037). Prognostic significance was not found in association with PD-L1 expression in PSC.
Discussion
This study revealed the following observations about PSC: (1) an intertumoral heterogenic but characteristic genetic landscape represented by non-recurrent TP53 mutations and frequent MET and KEAP1 mutations, (2) intratumoral heterogeneity in the alteration of EMT-related genes and CNV, (3) decreased APOBEC signatures during metastatic progression, (4) subclonal diversity of primary and metastatic tumors, and (5) strong PD-L1 expression.
In this study, we demonstrate that PSC have a highly diverse and heterogeneous molecular profile. Even for the most commonly mutated TP53 gene, the mutation sites varied in all tumors. Approximately one-third of primary PSC have oncogenic driver mutations found in NSCLC, such as EGFR (29%), KRAS (6%), and MET exon 14 skipping mutations (19%). TP53 mutations co-occurred with EGFR and/or KRAS mutations but not with MET exon 14 skipping mutations. In contrast to previous reports on PSC (EGFR 0%-8% and KRAS 22%-56%), we observed higher EGFR and lower KRAS mutation rates [6-9,20,21], which might be attributable to ethnic differences.
PSC was distinguishable from other NSCLC by its unique characteristics, such as more frequent MET and KEAP1 mutations and stronger PD-L1 expression. This study demonstrated that PSC harbored higher MET mutations (19%) than that reported in other NSCLC (~3%), which is parallel to previous reports [22,23]. In addition, we identified a novel frameshift deletion mutation (c.2901_2911del:p. E967fs), which leads to MET exon 14 skipping confirmed by ddPCR.
The results indicated that occurrence of MET exon 14 skipping mutations and MDM2 amplification and that of TP53 and other driver mutations were mutually exclusive. MET is a transmembrane receptor tyrosine kinase that activates various signaling pathways, promoting cell growth, survival, migration, and proliferation. MET exon 14 skipping mutation impairs ubiquitin-dependent degradation of MET, resulting in overexpression of MET protein, which in turn promotes tumor development and progression in cancer. MDM2 is associated with PI3K/AKT pathway, a signaling pathway activated by MET. MDM2, being stimulated by AKT, promotes cell survival inducing the ubiquitin-dependent degradation of p53. In this study, tumors with MET alteration frequently harbored MDM2 amplification. Therefore, two different mechanisms can stimulate MDM2 overexpression in these tumors, significantly downregulating p53. The overall effect may be similar to the consequences of the inactivation of TP53. Patients harboring MET exon 14 skipping mutations, including those with PSC, are eligible for targeted therapy with c-MET inhibitors, such as crizotinib or capmatinib [8,23].
In our study, 61.3% of PSC showed strong PD-L1 expression (TPS > 50). Recent evidence proposes a significant role of PD-L1 in EMT in various cancers, including LUAD [24,25]. Also, high PD-L1 expression is associated with elevated EMT markers, such as ZEB1, in adenocarcinoma cells. The higher PD-L1 expression in TSD compared with that in TCD, as observed in our study, supports the view that PD-L1 is possibly associated with EMT progression in PSC. However, we could not find any association between mutations in EMT genes and PD-L1 expression.
In addition, we demonstrated intratumoral heterogeneity of EMT-related gene alterations and CNV between TCD and TSD. The chromosomal region of 1q21.3, amplified more frequently in TSD than in TCD, harbors S100A and IRAK1, which are associated with tumor recurrence and chemoresistance in cancer [26]. These findings indicate that the sarcomatous part of PSC might represent tumor progression of carcinomatous component. Further transcriptomic analyses of EMT genes would confirm EMT-related genetic alterations and clarify the association of EMT with PD-L1 expression.
The APOBEC mutational signature is characteristically absent or reduced during the metastatic progression of PSC. The APOBEC mutational signature is the change of cytosine to thymine or guanine, attributed to the activity of the AID/APOBEC family of cytidine deaminases, especially APOBEC1, APOBEC3A, and/or APOBEC3B. These cytidine deaminases contribute to the subclonal expansion and intratumoral heterogeneity in the progression of NSCLC, particularly in its late stages [26,27]. Therefore, the decreased APOBEC signature observed in metastatic tumors of PSC is a distinctive feature that stands out from other NSCLC. Exploring the role of APOBEC signature in the progression of PSC would be an intriguing topic for future studies.
Recently, the APOBEC signature has been reported as a prospective predictor of immunotherapeutic response in NSCLC; furthermore, NSCLC patients with enriched APOBEC signature showed durable clinical benefit [27,28]. Unfortunately, since PSC loses its APOBEC signature during progression, despite high PD-L1 expression, advancedstage patients with PSC may be resistant to immunotherapy. However, since the APOBEC mutagenesis takes part in the development of drug resistance to tyrosine kinase inhibitors [29,30], patients with PSC may be more eligible to targeted therapies in late stages than patients with LUAD or LUSC.
In this study, the clinical outcomes, including 1-year and 5-year survival rates, were discovered to be better than previous reports [20], which may be attributable to the high proportion of patients at early stage present in the cohort. Moreover, it is difficult to interpret the lower TMB in metastatic lesions than in primary tumors of PSC, as detected in this study. This may be due to the low tumor purity of metastatic samples from lymph nodes.
There are a few limitations in this study. The small number and variety of metastatic samples included in this study restricted the thorough evolutionary analysis of PSC progression. The metastatic samples enrolled in the study were mostly associated with lymph node metastases; therefore, their mutational profile of should be interpreted according to lymphatic metastasis rather than hematogenous spread. Also, tumor purity in some metastatic and primary samples were as low as 15%, which may have influenced the results (S8 Table). Moreover, analysis on the RNA level was unattainable due to tissue insufficiency of most MD and some TSD samples. This limited the investigation on genetic translocations, the EMT signature, or signaling pathway analyses that could have revealed interesting results on tumor progression or PD-L1 expression.
Overall, PSC shows intertumoral and intratumoral heterogeneity in primary and metastatic tumors, which is mainly based on subclonal diversity. The clinical significance of the genetic mutations comprising this subclonal diversity is mostly unknown. On the other hand, clonal mutations with currently available targeted therapy, such as EGFR, were detected in every sample of the respective patients. Thus, repeated molecular testing on primary tumors would not be beneficial at this point. However, the high subclonal diversity seems to be associated with tumor progression in PSC, which may also affect patient’s treatment outcomes. Therefore, in the long run, additional testing may enable customized treatment strategies, ultimately leading to precision medicine.
The clinical aggressiveness and difficulty in treating PSC could be attributable to profound intratumoral and intertumoral histo-genetic diversity. Therefore, a single treatment strategy may not be sufficient to effectively control PSC. Next-generation sequencing would effectively aid the identification of the appropriate treatment strategy for individual tumors in PSC.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
This study was approved by the Institutional Review Board from Seoul National University Hospital (X-2303-819-903). Informed consent was waived because of the retrospective nature of this study.
Author Contributions
Conceived and designed the analysis: Kwon HJ, Chung JH.
Collected the data: Kwon HJ, Han YB, Lee J, Kwon S, Kim H.
Contributed data or analysis tools: Lee S, Chung JH.
Performed the analysis: Kwon HJ, Lee S, Han YB, Lee J, Kwon S, Kim H.
Wrote the paper: Kwon HJ.
Supervision and Project administration: Chung JH.
Data curation and Visualization: Lee S.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIT) (No: 2022R1A5A6000840).