Recent Developments in the Therapeutic Landscape of Advanced or Metastatic Hormone Receptor–Positive Breast Cancer
Article information

Abstract
Hormone receptor–positive (HR+) disease is the most frequently diagnosed subtype of breast cancer. Among tumor subtypes, natural course of HR+ breast cancer is indolent with favorable prognosis compared to other subtypes such as human epidermal growth factor protein 2–positive disease and triple-negative disease. HR+ tumors are dependent on steroid hormone signaling and endocrine therapy is the main treatment option. Recently, the discovery of cyclin-dependent kinase 4/6 inhibitors and their synergistic effects with endocrine therapy has dramatically improved treatment outcome of advanced HR+ breast cancer. The demonstrated efficacy of additional nonhormonal agents, such as targeted therapy against mammalian target of rapamycin and phosphatidylinositol 3-kinase signaling, poly(ADP-ribose) polymerase inhibitors, antibody-drug conjugates, and immunotherapeutic agents have further expanded the available therapeutic options. This article reviews the latest advancements in the treatment of HR+ breast cancer, and in doing so discusses not only the development of currently available treatment regimens but also emerging therapies that invite future research opportunities in the field.
Introduction
Breast cancer is the most commonly diagnosed cancer worldwide. It is a leading cause of death among women worldwide, with an estimated 2.3 million new cases each year and accounting for over 680,000 deaths in 2020 [1].
Breast cancer is not a single disease entity. Microarray-based gene expression profiling has identified four intrinsic subtypes of breast cancer, which include luminal A, luminal B, human epidermal growth factor protein 2 (HER2)–overexpression, and triple-negative breast cancer (TNBC) [2]. Each subtype also corresponds to a distinct histopathological profile based on the expression of hallmark biomarkers, namely the hormone receptors (HR) for estrogen (ER) or progesterone, HER2, and the cell proliferation marker Ki67. Briefly, luminal A tumors are HR+, HER2–, and Ki67-low; luminal B tumors are further subdivided into HR+/HER2–/ Ki67-high and HR+/HER2+/Ki67-low subtypes; HER2- overexpression refers to HR–/HER2+ tumors; and TNBC is negative for both HR and HER2 [3]. The luminal A and B subtypes are collectively referred to as HR+ breast cancer, which represents over two-thirds of all breast cancer diagnoses and has a more favorable prognosis compared to TNBC [4]. This can be attributed to not only its natural history but also its dependence on estrogen signaling, which is targeted with endocrine therapy agents such as selective ER modulators (SERMs), aromatase inhibitors (AIs), and selective ER degraders (SERDs) [5].
While endocrine therapy continues to be a mainstay of treatment for HR+ breast cancer, the discovery of additional nonhormonal molecular targets—namely cyclin-dependent kinase 4/6 (CDK4/6), mammalian target of rapamycin (mTOR), protein kinase B (also known as Akt), and phosphatidylinositol 3-kinase (PI3K)—and their translation into novel therapeutic agents have further improved patient outcomes. Despite these milestones, advanced HR+ breast cancer remains an incurable condition, often with mechanisms independent of hormonal signaling that contribute to recurrence, therapeutic resistance, and/or metastasis, underscoring the need for further translational research in this realm. In this review, we will discuss the currently available and emerging therapeutic agents in advanced HR+ breast cancer, highlighting landmark studies that have paved their introduction into clinical practice.
Endocrine Therapy Plus CDK4/6 Inhibitors as First-Line Treatment for Advanced HR+ Breast Cancer
Endocrine therapy is the main treatment option for advanced HR+ breast cancer. Endocrine therapy can be broadly classified into SERMs, AIs, and SERDs. SERMs are competitive inhibitors of estrogen-ER binding and display either agonist or antagonist activity depending on the target tissue; for instance, tamoxifen, the most commonly used SERM, has anti-proliferative effects in breast tissue but agonistic effects in the endometrium, bone, and cardiovascular tissues [6]. AIs inhibit the conversion of androgens to estradiol in non-ovarian tissues and are used to reduce estrogen levels in the postmenopausal setting. Nonsteroidal AIs, which include anastrozole and letrozole, are noncovalent, reversible competitive inhibitors of androgen-aromatase binding, whereas the steroidal AI exemestane covalently and irreversibly binds to the aromatase substrate-binding site [7]. SERDs such as fulvestrant are a more recently developed class of endocrine agents that antagonize ER signaling and result in ER degradation. Endocrine therapy is effective in most patients with HR+ advanced breast cancer. However, cancer cells acquire new mutations or lose HR expression, developing endocrine therapy resistance over time [5]. Traditionally, primary endocrine resistance is defined as patients who relapse during the first 2 years of adjuvant endocrine therapy or disease progression within the first 6 months of first-line endocrine therapy for advanced or metastatic breast cancer. Secondary (acquired) resistance is defined as relapse during adjuvant endocrine therapy but after the first 2 years, relapse within 12 months of completing adjuvant endocrine therapy or disease progression 6 months after endocrine therapy for advanced or metastatic breast cancer [8].
In addition to endocrine therapy, the discovery of CDK4/6 inhibitors has further transformed the treatment landscape for advanced HR+ breast cancer. CDK4 and CDK6 are serinethreonine kinases that facilitate G1/S progression in the cell cycle by inhibiting the retinoblastoma pathway, and studies have shown that their activation contributes to an ER-independent growth signal in HR+ breast cancers [9]. In 2009, the CDK4/6 inhibitor, palbociclib was shown to exhibit growth inhibitory effects on ER+ breast cancer cell lines in vitro [10]. This included both synergy with tamoxifen as well as potency against cell lines with tamoxifen resistance, inviting a novel therapeutic opportunity for HR+ breast cancers refractory to endocrine therapy [10]. Subsequent drug discovery efforts also led to the development of ribociclib and abemaciclib; today, all three CDK4/6 inhibitors are approved by the U.S. Food and Drug Administration (FDA) and Ministry of Food and Drug Safety (MFDS) in Korea as first-line treatment for advanced HR+ breast cancer combined with endocrine therapy. Landmark studies of CDK4/6 inhibitors are summarized in Table 1 [11-26].
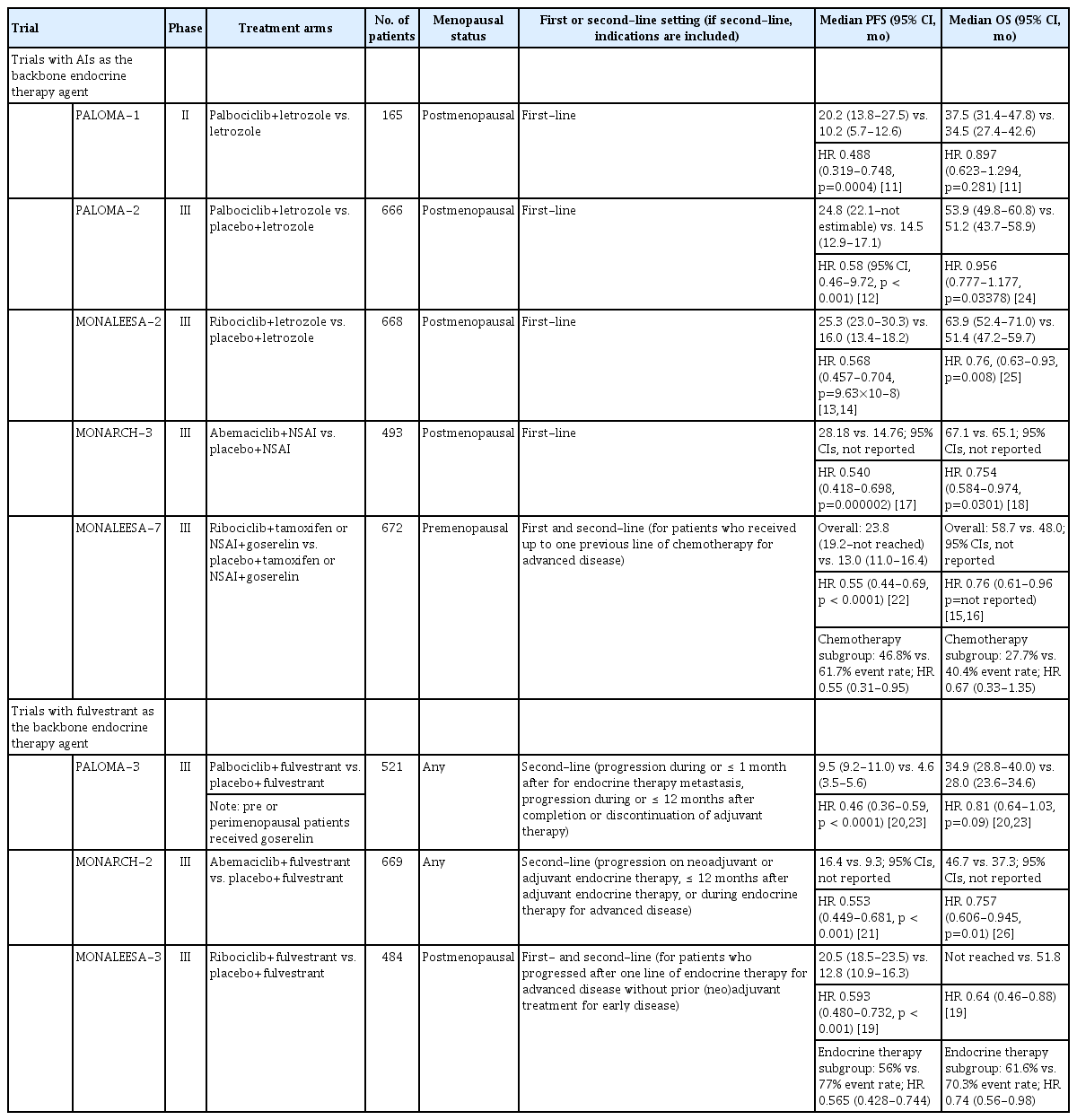
Landmark studies in dual CDK4/6 inhibitor and endocrine therapy for advanced HR+breast cancer
The phase II PALOMA-1 study demonstrated that firstline palbociclib and letrozole combination therapy significantly increased progression-free survival (PFS) compared to letrozole monotherapy in postmenopausal patients with advanced ER+/HER2– breast cancer (20.2 vs. 10.2 months; hazard ratio, 0.488; p=0.0004) [11]. These results, which were further confirmed by the phase III PALOMA-2 trial [12], led to accelerated U.S. FDA approval of palbociclib in combination with letrozole for ER+/HER2– breast cancer in 2015. The MONALEESA-2 trial showed increased PFS with ribociclib/letrozole compared to letrozole alone (25.3 vs. 16.0 months; hazard ratio, 0.568; p=9.63×10–8) [13,14], and MONALEESA-7 was the first study to demonstrate overall survival (OS) benefits for adding ribociclib to therapy with goserelin and either letrozole or tamoxifen (58.7 vs. 48.0 months; hazard ratio, 0.76; 95% confidence interval [CI], 0.61 to 0.96) [15,16] in premenopausal women. The PFS benefits of abemaciclib in combination with either anastrozole or letrozole relative to AI plus placebo was first demonstrated in the MONARCH-3 trial (28.2 vs. 14.8 months; hazard ratio, 0.540; p=0.000002) [17,27]. OS was also reported to be significantly prolonged in the abemaciclib arm (67.1 vs. 65.1 months; hazard ratio, 0.754; p=0.0301) [18].
The MONALEESA-3 trial investigated the efficacy of ribociclib plus fulvestrant as first-line or second-line treatment in advanced HR+ breast cancer patients. Around half of patients were treatment naïve for advanced disease and prior chemotherapy for advanced disease was not allowed. Overall, patients in the ribociclib plus fulvestrant arm showed prolonged PFS (20.5 vs. 12.8 months; hazard ratio, 0.593; p < 0.001) and OS (53.7 vs. 41.5 months; hazard ratio, 0.73; 95% CI, 0.59 to 0.90) relative to those in the control arm [19,28]. Subgroup analysis of those who had not received endocrine therapy in the advanced setting (treatment naïve patients) also showed prolonged PFS (32% vs. 51% event rate; hazard ratio, 0.577; 95% CI, 0.415 to 0.802) and OS (34.5% vs. 52.7% deaths; hazard ratio, 0.62; 95% CI, 0.41 to 0.95) in the ribociclib plus fulvestrant arm [19,28].
As a result, combined treatment with endocrine therapy and CDK4/6 inhibitors is recommended as first-line therapy for advanced HR+ breast cancer [8,29]. CDK4/6 inhibitors are effective in de novo or recurrent metastatic breast cancer, and in patients with primary or secondary endocrine resistance [8]. Although chemotherapy was previously regarded as first-line therapy in the setting of visceral crisis, studies are underway to evaluate the safety and efficacy of CDK4/6 inhibitors in this setting as well. Most recently, preliminary results from the phase II RIGHT choice trial reported a statistically significant PFS benefit among premenopausal advanced HR+/HER2– breast cancer patients with 10% or higher expression of ER by immunohistochemistry and aggressive features (rapidly progressing or highly symptomatic disease, including visceral crises) in the ribociclib+letrozole/anastrazole+goserelin arm relative to those receiving chemotherapy (24.0 vs. 13.0 months; hazard ratio, 0.54; p=0.0007) [30].
With regards to the preferred CDK4/6 inhibitor for treatment, the preferred agent is decided on a case-by-case basis based on patient risk factors for adverse events, as well as PFS and/or OS benefits seen in pivotal clinical trials [31]. Neutropenia is a common adverse effect of all three CDK4/6 inhibitors, though it is more frequently observed with palbociclib and ribociclib. For instance, across the three trials that investigated dual therapy with fulvestrant and a CDK4/6 inhibitor, the incidence of all grade neutropenia with palbociclib, ribociclib, and abemaciclib were 78.8%, 69.6%, and 46.0%, respectively [20,21,28]. This discrepancy has been attributed to the greater selectivity of abemaciclib for CDK4 over CDK6 [32,33]. Furthermore, unlike chemotherapy-induced neutropenia, neutropenia associated with CDK4/6 inhibition is rapidly reversible [32,33]. Diarrhea is more common with abemaciclib, which was observed in 86.4% of patients in the MONARCH-2 trial, whereas 19.1% and 29.0% of patients in the PALOMA-3 (palbociclib) and MONARCH-2 (ribociclib) trials reported diarrhea [20,21,28]. Overall, the consensus is that adverse events associated with CDK4/6 inhibitors are well managed with standard supportive therapy and when indicated, dose interruptions or reductions [32,33].
If the patients did not recur while on adjuvant nonsteroidal AI or within 12 months after completing therapy, AI plus CDK4/6 inhibitor is recommended. There was no clear benefit of fulvestrant plus CDK4/6 inhibitor over AI plus CDK4/6 inhibitor in a phase II study [34]. Likewise, AI plus CDK4/6 inhibitor is recommended in patients who had relapsed on adjuvant tamoxifen. Fulvestrant plus CDK4/6 inhibitors are recommended in patients who relapsed on adjuvant AI, or within 12 months after completing adjuvant AI therapy.
Endocrine Therapy Plus CDK4/6 Inhibitors as Second- or Subsequent-Line Treatment for Advanced HR+ Breast Cancer
Numerous studies have also shown the benefits of combined CDK4/6 inhibitor and endocrine therapy in the second-line or early relapse setting. In addition to demonstrating the benefits of combined ribociclib and endocrine therapy as first-line treatment for advanced HR+ breast cancer, the MONALEESA-7 trial also investigated the benefits of adding ribociclib in patients who received no more than one line of chemotherapy for metastasis. Around 14% of patients enrolled in the MONALEESA-7 trial received 1 line of chemotherapy for advanced disease. Compared to those who received goserelin and either letrozole or tamoxifen alone, those who received ribociclib, goserelin and endocrine therapy showed improved PFS (46.8% vs. 61.7%; hazard ratio, 0.55; 95% CI, 0.31 to 0.95) but not OS (53.2% vs. 65.9% deaths; hazard ratio, 0.747; 95% CI, 0.441 to 1.266) [16,22].
As for regimens with fulvestrant as the backbone, the PALOMA-3 trial was the first to investigate the efficacy of combined palbociclib and fulvestrant as second-line or later line of therapy for advanced HR+ breast cancer patients who progressed on prior endocrine therapy [20]. Patients who had received one prior line of chemotherapy in the advanced setting were also included. Specifically, patients on dual-palbociclib/fulvestrant therapy exhibited significantly longer PFS compared to the placebo/fulvestrant group (9.5 vs. 4.6 months; hazard ratio, 0.46; p < 0.0001) [23]. Additionally, patients who received one line of prior chemotherapy for metastasis exhibited prolonged PFS (7.7 vs. 3.5 months; HR, 0.43; 95% CI, 0.29 to 0.64). Subsequent analyses showed OS benefits when restricted to patients with sensitivity to previous endocrine therapy (39.7 vs. 29.7 months; hazard ratio, 0.72), although statistical significance was not reached in analyses of the entire trial group [35].
The MONARCH-2 trial also tested the efficacy of combined abemaciclib/fulvestrant therapy in patients who progressed while on or 12 months after neoadjuvant or adjuvant endocrine therapy, or while receiving endocrine therapy in the advanced setting. Since subgroup analyses were performed based on primary or secondary endocrine therapy resistance as defined by the European School of Oncology (ESO)/European Society of Medical Oncology (ESMO) guidelines [36], the specific PFS and OS values for patients who progressed on endocrine therapy for advanced breast cancer are unknown. Nonetheless, the overall cohort of individuals in the abemaciclib/fulvestrant arm exhibited prolonged PFS (16.4 vs. 9.3 months; hazard ratio, 0.553; p < 0.001) and OS (45.8 vs. 37.2 months; hazard ratio, 0.784; 95% CI, 0.644 to 0.955) compared to the placebo/fulvestrant arm [21,37]. For patients who received treatment as second-line therapy or who had early relapse MONALEESA-3 reported improved PFS (14.6 vs. 9.1 months; hazard ratio, 0.57; 95% CI, 0.44 to 0.74) and OS (40.2 vs. 32,5 months; hazard ratio, 0.73; 95% CI, 0.53 to 1.00) with dual ribociclib/fulvestrant therapy relative to placebo/fulvestrant [38].
Finally, abemaciclib is the only CDK4/6 inhibitor that is approved as monotherapy for treatment-refractory HR+/HER2– metastatic breast cancer. The phase II single-arm MONARCH-1 trial reported an overall response rate (ORR) of 19.7%, median PFS of 6.0 months, and a median OS of 17.7 months with abemaciclib monotherapy in a cohort where 90.2% of patients had visceral disease and 50.8% had at least three sites of metastasis [39]. More recently, the phase II nextMONARCH trial randomized patients with endocrinerefractory HR+/HER2– metastatic breast cancer who previously received chemotherapy to abemaciclib 150 mg+tamoxifen 20 mg (A+T), twice-daily abemaciclib 150 mg (A-150) monotherapy, or once-daily abemaciclib 200 mg+loperamide (A-200) for diarrhea prophylaxis. There were no significant PFS differences between A+T and A-150 (9.1 vs. 7.4 months; hazard ratio, 0.815; p=0.293) and between A-150 and A-200 (6.5 vs. 7.4 months; hazard ratio, 1.045; p=0.811) [40].
Endocrine Therapy Combined with Agents Targeting the PI3K/AKT/mTOR Pathway
There is growing evidence that aberrant activation of the PI3K/Akt/mTOR pathway in association with ER signaling underlies acquired resistance to endocrine therapy in HR+ breast cancer [41]. Studies have reported somatic gain-of-function mutations in PIK3CA, which encodes the tyrosine kinase PI3K, in approximately 40% of patients with HR+ breast cancer [42]. While CDK4/6 inhibitor combined with endocrine therapy is an initial treatment option for advanced HR+ breast cancer, there are additional agents for subsequent treatment targeting the PI3K/Akt/mTOR pathway. Current guidelines for this population recommend combination therapy with fulvestrant, a SERD, and the PI3K inhibitor, alpelisib as second-line therapy [8,31]. The SOLAR-1 trial is a phase III randomized controlled trial that compared fulvestrant with alpelisib to fulvestrant with placebo in HR+ breast cancer. Patients with PIK3CA mutations who received fulvestrant and alpelisib had prolonged PFS (11.0 vs. 5.7 months; hazard ratio, 0.65; p < 0.001) and ORR, (26.6% vs. 12.8%) compared to those who received fulvestrant and placebo [43]. The combination regimen did not significantly prolong PFS nor ORR in individuals without PIK3CA mutations. In the subsequent BYLieve trial, the efficacy of alpelisib plus fulvestrant was maintained in PIK3CA-mutated HR+ breast cancer, who had progression on a CDK4/6 inhibitor plus an aromatase inhibitor [44]. These results led to U.S. FDA and Korean MFDS approval of fulvestrant and alpelisib combination therapy in advanced HR+ breast cancer with PIK3CA mutations. However, alpelisib is not without its safety risks; hyperglycemia was one of the most common adverse effects in the SOLAR-1 trial with an incidence of 63.7% in the alpelisib/fulvestrant arm compared to 9.8% in the placebo/fulvestrant arm [43]. Of note, individuals with known type 1 diabetes or uncontrolled type 2 diabetes (hemoglobin A1c > 6.4% or fasting plasma glucose > 140 mg/dL) were excluded from the study. Other common side effects included gastrointestinal toxicities (i.e., diarrhea in 57.7% and 15.7%, respectively, nausea in 44.7% and 22.3%, decreased appetite in 35.6% vs. 10.5%) and rash (35.6% and 5.9%).
More recently, the phase II FAKTION trial reported that combination therapy with capivasertib, an AKT inhibitor, and fulvestrant prolonged PFS (10.3 vs. 4.8 months; hazard ratio, 0.56; p=0.0023) and OS (29.3 vs. 23.4 months; hazard ratio, 0.66; p=0.035) relative to placebo/fulvestrant in advanced HR+/HER2– breast cancer with relapse or progression on an AI [45,46]. A phase III CAPItello-291 trial investigated the safety and efficacy of this dual regimen in patients with AI-resistant advanced HR+/HER2– breast cancer, notably including those who have previously received CDK4/6 inhibitors [47]. PFS was significantly prolonged in both the overall cohort (7.2 vs. 3.6 months; hazard ratio, 0.60; p < 0.001) and in patients with genetic alterations in the AKT pathway (7.3 vs. 3.1 months; hazard ratio, 0.50; p < 0.001) [47].
Everolimus, an mTOR inhibitor, has also been investigated for advanced HR+ breast cancer previously treated with nonsteroidal AI. The phase III BOLERO-2 trial demonstrated improved PFS in advanced HR+ breast cancer patients on exemestane in conjunction with everolimus compared to exemestane alone (7.8 vs. 3.2 months; hazard ratio, 0.45; p < 0.0001) [48,49]. Although there was no significant improvement in OS [50], the combination therapy was approved for metastatic HR+ breast cancer that progressed on an AI in 2012. Subsequent analyses on the cell-free DNA for activating mutations in the ER-encoding ESR1 gene found that while patients with ESR1 mutations overall exhibited shorter PFS compared to those with wild-type ESR1, those with D538G mutations who received exemestane and everolimus demonstrated a similar PFS to wild-type individuals [51]. These results highlight the relationship between ER and mTOR signaling, as well as the utility of circulating biomarkers for monitoring therapeutic response and prognosis.
Oral SERDs as Backbone Endocrine Therapy Agents in Advanced HR+ Breast Cancer
Despite the efficacy of fulvestrant as a backbone endocrine therapy agent in combined treatment regimens for advanced HR+ breast cancer, it is available only via intramuscular injection due to its poor oral bioavailability, which imposes a practical barrier to its long-term use. As a result, there have been considerable efforts to develop orally available SERDs such as elacestrant, which demonstrated improved PFS over standard-of-care endocrine monotherapy in the phase III EMERALD trial (6-month PFS rates 34.3% vs. 20.4%; 12-month PFS rates 22.3% vs. 9.4%; HR, 0.70; p=0.0018) [52]. Importantly, the trial enrolled patients with advanced HR+/ HER2– breast cancer who progressed on previous dual therapy with a CDK4/6 inhibitor and fulvestrant or an AI, and PFS benefits were also observed among patients with ESR1 mutations (6-month PFS rates 40.8% vs. 19.1%; 12-month PFS rates 26.8% vs. 8.2%; hazard ratio, 0.55; p=0.0005). Moreover, improved PFS was reported when elacestrant was specifically compared against fulvestrant in both the overall cohort (6-month PFS rates 40.8% vs. 20.8%; 12-month PFS rates 26.8% vs. 8.4%; hazard ratio, 0.50; p=0.0005) and in patients with altered ESR1 (6-month PFS rates 34.3% vs. 22.9%; 12-month PFS rates 22.3% vs. 10.2%; p=0.0049). In 2023, elacestrant was FDA-approved for postmenopausal women or adult men with HR+/HER2–, ESR1-mutated advanced or metastatic breast cancer who progressed on at least one line of endocrine therapy.
In addition to elacestrant, many oral SERDs are under investigation [53]. In patients with HR+/HER2– breast cancer who progressed after at least one line of endocrine therapy, camizestrant improved PFS at both the 75 mg (7.2 vs. 3.7 months; hazard ratio, 0.58; 95% CI, 0.41 to 0.81; p=0.0124) and 150 mg (7.7 vs. 3.7 months; hazard ratio, 0.67; 95% CI, 0.48 to 0.92; p=0.0161) doses compared to fulvestrant in the SERENA-2 trial [54]. The phase III SERENA-4 and SERENA-6 trials are currently enrolling patients to investigate the relative efficacy of combination therapy with a CDK4/6 inhibitor and either camizestrant or an AI [55,56]. In the acelERA BC trial which included HR+/HER2– breast cancer who progressed after at least one line of endocrine therapy, although the study did not meet its primary endpoint, giredestrant showed numerical improvement of PFS compared to physician choice of endocrine monotherapy especially in those with ESR1 mutation (5.3 vs. 3.5 months; hazard ratio, 0.60; 95% CI, 0.35 to 1.03, p=0.061) [57]. Finally, newer classes of agents that target the ER, including proteolysis targeting chimeras (PROTACs), selective ER covalent antagonists (SERCAs), and complete ER antagonists (CERANs), are currently being developed and tested for use in both the early and advanced settings [53]. Briefly, PROTACs simultaneously bind to the ER and an E3 ubiquitin ligase for ubiquitinmediated proteasomal degradation of the ER; SERCAs result in ER inactivation via a covalent bond to a key cysteine residue of wildtype and mutant ER proteins; CERANs inhibit ER-mediated transcriptional activation [58,59].
Emerging Non-endocrine Therapies in Advanced HR+ Breast Cancer
1. Poly(ADP-ribose) polymerase inhibitors for BRCA1/2-mutant cancer
Perhaps the most well-established genetic factor for breast cancer are pathogenic mutations in BRCA1/2, which are tumor suppressor genes that encode DNA repair enzymes involved in homologous recombination. Briefly, the recognition of a double-strand break (DSB) leads to the phosphorylation of BRCA1 as well as other downstream targets, which together act as a scaffold for the complex machinery that mediates DSB repair [60]. The ends of damaged DNA are then resected to form single-strand templates for repair, after which BRCA2, in a complex with additional DNA repair proteins, loads the critical RAD51 protein onto the site of damage for strand invasion and homologous recombination [60]. Pathogenic BRCA1/2 mutations are highly penetrant and significantly increase lifetime risk of breast cancer in an autosomal dominant fashion, accounting for approximately 5%-10% of breast cancers [61,62]. Another key family of DNA repair enzymes are poly(ADP-ribose) polymerase (PARP) proteins, which facilitate single-strand break (SSB) repair as well as base excision repair. Suppression of PARP activity leads to the accumulation of unresolved SSBs that degrade into DSBs, which in BRCA1/2-mutant cells that are incapable of HR, results in genomic instability and cell death [61]. This phenomenon has been leveraged in the development of PARP inhibitors as therapeutic agents for breast cancer. The phase III OlympiAD trial demonstrated the efficacy of monotherapy with the PARP inhibitor olaparib compared to single-agent chemotherapy in HER2– metastatic breast cancer patients with a germline BRCA1/2 mutation [63]. Significantly prolonged PFS was reported in the olaparib arm (7.0 vs. 4.2 months; hazard ratio, 0.58; p < 0.01), and while there was no significant improvement in OS (19.3 vs. 17.1 months; hazard ratio, 0.90; p=0.513), subgroup analyses suggested potential benefit in individuals who had not received prior chemotherapy in the metastatic setting [63,64]. Similarly, talazoparib also exhibited significantly prolonged PFS (8.6 vs. 5.6 months; hazard ratio, 0.54; p < 0.001) and increased ORR (62.6% vs. 27.2%; odds ratio, 5.0; p < 0.001) in germline BRCA1/2 mutant advanced breast cancer relative to singleagent chemotherapy in the phase III EMBRACA trial [65]. Both medications have been approved for germline BRCA1/2 mutant advanced breast cancer as of 2014 and 2018, respectively. Consideration for monotherapy with PARP inhibitors is also recommended for patients with pathogenic variations in PALB2, which encodes a protein that co-localizes with and stabilizes BRCA2 to mediate RAD51 loading for homologous recombination [8].
2. HER2-directed therapy for HER2-low cancers
The development of HER2-directed therapy is considered one of the greatest advancements in breast cancer treatment, and trastuzumab, a monoclonal anti-HER2 antibody, is a well-established agent for the treatment of HER2+ breast cancer. Nonetheless, there is growing evidence for HER2- therapy in metastatic HR+ tumors with low expression of HER2 (HER2 1+ or 2+ on immunohistochemistry and negative on fluorescence in situ hybridization), which account for 45%-55% of breast cancers [66]. Trastuzumab-deruxtecan (T-Dxd) is an antibody-drug conjugate (ADC) in which trastuzumab is linked to the topoisomerase I inhibitor deruxtecan. Upon its release into the HER2-expressing target cell, the deruxtecan “payload” portion of T-DXd exerts a potent, cytotoxic “bystander effect” on surrounding cells, including HER2– cells [67]. A phase Ib study of T-DXd demonstrated antitumor effects in patients with advanced HER2-low breast cancer refractory to standard therapy (median duration of response 10.4 months; 95% CI, 8.8 months to not evaluable) [68]. Following this, the DESTINY-Breast04 phase III randomized clinical trial demonstrated that T-DXd significantly prolonged both PFS (10.1 vs. 5.4 months; hazard ratio, 0.51; p < 0.001) and OS (23.9 months vs. 17.5 months; hazard ratio, 0.64; p=0.003) compared to the physician’s choice of chemotherapy [69]. These results held true not just for the HR+ group, but also among all enrolled patients regardless of HR+ disease, and led to U.S. FDA approval of T-DXd for advanced HER2-low breast cancer.
3. Sacituzumab govitecan
Sacitizumab-govitecan is another ADC that is being actively investigated for use in HR+ tumors. Recently approved for metastatic TNBC, the ADC consists of an anti-Trop2 antibody and a topoisomerase I payload. Trop2, which is overexpressed in a variety of malignant tumors including invasive breast cancer, is a transmembrane glycoprotein that upregulates proliferative markers while downregulating pro-apoptotic proteins to ultimately promote tumor growth [70]. A phase I/II trial of 54 patients with metastatic HR+/HER2– who progressed despite endocrine therapy and received at least one round of chemotherapy in the metastatic setting demonstrated encouraging results with an ORR of 31.5% (95% CI, 19.5 to 45.6) and a PFS of 5.5 months (95% CI, 3.6 to 7.6) [71]. Recently, a phase III TROPiCS-02 trial demonstrate an improved PFS (5.5 vs. 4.0 months; HR, 0.66; p=0.0003) and OS (14.4 vs. 11.2 months; HR, 0.79; p=0.020) with sacituzumab-govitecan compared to conventional chemotherapy in patients with relapsed or refractory HR+/HER2– breast cancer [72,73].
4. Immunotherapy in advanced HR+ breast cancer
In line with recent breakthroughs in immunotherapy for solid tumor malignancies, there are increasing efforts to adopt immune checkpoint inhibitors into available therapeutic agents for breast cancer. Immune checkpoint inhibitors were initially introduced in the setting of TNBCs, since HR+ tumors have been traditionally described as “immunologically cold” with low numbers of tumor-infiltrating lymphocytes. The limited efficacy of the anti-PD1 antibody pembrolizumab in the phase Ib KEYNOTE-028 trial, which enrolled metastatic HR+/HER2– breast cancer patients with >1% programmed death-ligand 1 (PD-L1)–expressing stroma or tumor cells (ORR, 12%; 95% CI, 2.5 to 31.2; no complete responses) [74], has been attributed to this finding; however, there is active research into the mechanisms underlying the architecture of the tumor microenvironment in HR+ breast cancer and ways to increase its susceptibility to cancer immunotherapy [75]. Similarly, initial trials testing the combination of pembrolizumab with chemotherapeutic agents such as eribulin or capecitabine did not demonstrate significant PFS or ORR benefits [76,77]. However, a more recent single-arm phase II trial of pembrolizumab/ eribulin therapy in metastatic HR+ breast cancer reported promising results (median PFS of 6.0 months; 95% CI, 3.7 to 8.4; 1-year OS, 59.1%; 95% CI, 45.8 to 76.2), which authors attributed to the degree of pre-treatment with chemotherapy and differences in PD-L1 scoring methods [78]. In addition, nivolumab plus eribulin showed ORR of 53.3% and median PFS of 5.6 months (95% CI, 4.3 to 6.8) in a phase IB/II study of HR+HER2– patients [79]. The currently ongoing KEYNOTE-B49 phase III trial is also evaluating the comparative efficacy of pembrolizumab in combination with one of four chemotherapy regimens, paclitaxel, nab-paclitaxel, doxorubicin, or capecitabine [80]. Additionally, a phase I/II trial of triple therapy with palbociclib, pembrolizumab, and letrozole in metastatic HR+/HER2– breast cancer observed 31% and 25% complete and partial response rates, inviting further research into the potential synergistic effects of immune checkpoint therapy with CDK4/6 inhibitors [81]. The role for immune checkpoint inhibitor-based therapeutic strategies in advanced HR+ breast cancer therefore remains an open question that warrants continued investment to maximize their potential benefits.
In summary, both existing and emerging therapeutic agents for advanced HR+ breast cancer serve as a testament to the wealth of recent scientific and clinical developments that have transformed disease management, prognosis, and patient outcomes (Fig. 1). In turn, these discoveries have fueled further research into disease biology that ensure the continued evolution of available treatment regimens for advanced HR+ breast cancer. Key unanswered questions include the identification of biomarkers for disease monitoring, response to targeted therapy, and/or resistance, optimization of combined treatment regimens—particularly those that prolong OS—and the minimization of treatment-related toxicity.

Treatment of advanced/metastatic hormone receptor (HR)–positive, human epidermal growth factor protein 2 (HER2)–negative breast cancer. ESR1, estrogen receptor 1; ET, endocrine therapy; OFS, ovarian function suppression; PD, progressive disease; SERD, selective estrogen receptor degrader; T-Dxd, trastuzumab-deruxtecan; ULN, upper limit of normal.
Notes
Author Contributions
Wrote the paper: Lee EY, Lee DW, Lee KH, Im SA.
Revision: Lee EY, Lee DW, Lee KH, Im SA.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.