First-in-Human Phase 1 Study of a B Cell– and Monocyte-Based Immunotherapeutic Vaccine against HER2-Positive Advanced Gastric Cancer
Article information

Abstract
Purpose
BVAC-B is an autologous B cell– and monocyte-based immunotherapeutic vaccine that contains cells transfected with a recombinant human epidermal growth factor receptor 2 (HER2) gene and loaded with the natural killer T cell ligand alpha-galactosylceramide. Here, we report the first BVAC-B study in patients with HER2-positive advanced gastric cancer.
Materials and Methods
Patients with advanced gastric cancer refractory to standard treatment with HER2+ immunohistochemistry ≥ 1 were eligible for treatment. Patients were administered low (2.5×107 cells/dose), medium (5.0×107 cells/dose), or high dose (1.0×108 cells/dose) of BVAC-B intravenously four times every 4 weeks. Primary endpoints included safety and maximum tolerated BVAC-B dose. Secondary endpoints included preliminary clinical efficacy and BVAC-B-induced immune responses.
Results
Eight patients were treated with BVAC-B at low (n=1), medium (n=1), and high doses (n=6). No dose-limiting toxicity was observed, while treatment-related adverse events (TRAEs) were observed in patients treated with medium and high doses. The most common TRAEs were grade 1 (n=2) and grade 2 (n=2) fever. Out of the six patients treated with high-dose BVAC-B, three had stable disease with no response. Interferon gamma, tumor necrosis factor-α, and interleukin-6 increased after BVAC-B treatment in all patients with medium and high dose, and HER2-specific antibody was detected in some patients.
Conclusion
BVAC-B monotherapy had a safe toxicity profile with limited clinical activity; however, it activated immune cells in heavily pretreated patients with HER2-positive gastric cancer. Earlier treatment with BVAC-B and combination therapy is warranted for evaluation of clinical efficacy.
Introduction
Gastric cancer is the fifth most common cancer and the third leading cause of cancer-related deaths worldwide [1]. Human epidermal growth factor receptor 2 (HER2), which is associated with cell proliferation, migration, and differentiation, is an important and established target in gastric cancer [2]. Currently, the combination of trastuzumab, a monoclonal antibody against HER2, with fluoropyrimidine and a platinum compound is the standard first-line palliative treatment for gastric cancer patients with HER2 gene amplification [3]. Despite advancements in HER2-targeted treatments, many agents, including trastuzumab emtansine, lapatinib, and pertuzumab, that have shown positive results in breast cancer patients, did not improve survival in gastric cancer patients [4].
Recently, trastuzumab deruxtecan showed clinical activity in patients with HER2-positive gastric cancer who progressed after prior trastuzumab treatment [5]. Additional drugs include ZW25, a bispecific antibody against two HER2 epitopes, and margetximab that is used in clinical trials to investigate the clinical efficacy in HER2-positive gastric cancer, although final results are still pending. Currently, only trastuzumab deruxtecan is effective in HER2-positive gastric cancer patients that progressed after trastuzumab treatment; therefore, more treatment options for HER2-positive gastric cancer patients after failure of trastuzumab plus chemotherapy are required [6].
Investigators have focused on pathways involved in immune evasion, one of the key hallmarks of cancer, by applying programmed death-1 (PD-1) checkpoint inhibitors, such as pembrolizumab and nivolumab that show durable efficacy in multiple solid tumors including gastric cancer [7,8]. Despite durable responses in a subset of patients with both immunotherapeutic agents, the overall response rate (ORR) of anti–PD-1 monotherapy is only 10%–15%. Checkmate 649 showed that the combination of nivolumab with chemotherapy as first-line treatment in HER2-negative gastric cancer patients showed promising results with overall survival (OS) benefits [9]. Adding pembrolizumab to standard treatment with trastuzumab and chemotherapy in first-line settings for HER2-positive gastric adenocarcinoma patients in a phase 2 study showed 70% progression-free survival (PFS) at 6 months and 91% ORR [10]. Furthermore, a randomized phase 3 trial that compared pembrolizumab, trastuzumab plus chemotherapy, trastuzumab, and chemotherapy alone, reported improved ORR in the preliminary interim analysis [11].
The unmet need to target both HER2 and pathways of immune evasion has led to the development of adoptive cell therapies and vaccines against HER2 [12]. Chimeric antigen receptor (CAR) T cell therapy against CD19-expressing tumors demonstrated promising efficacy in hematological malignancies [13]; however, the efficacy of CAR T cell therapy against solid cancers showed marginal efficacy because of the lack of tumor-specific antigens (TSAs) and the presence of an immune-inhibitory tumor microenvironment (TME) [14]. Sipuleucel-T was the first U.S. Food and Drug Administration–approved cell-based immunotherapy for metastatic prostate cancer, and various tumor peptide vaccines such as G17DT, vascular endothelial growth factor receptor, and OTSTGC-A24 have been investigated for the treatment of gastric cancer [14].
An alternative cell-based immunotherapy, B cell– and monocyte-based anti-tumor vaccine therapy, has also been investigated [15]. BVAC-C is the first-in-class B cell– and monocyte-based immunotherapeutic vaccine against cervical cancer, which makes use of autologous B cells and monocytes as antigen presenting cells for the viral oncogenes E6 and E7 of human papillomavirus (HPV) types 16 and 18. The first human study of BVAC-C demonstrated durable anti-tumor activity in patients with HPV16/18 positive cervical cancer [16]. We developed BVAC-B, a B cell– and monocyte-based immunotherapeutic vaccine using cells transfected with recombinant HER2 and loaded with the natural killer T (NKT) cell ligand alpha-galactosylceramide. Preclinical models have shown that BVAC-B elicited immune responses by increasing the production of interferon γ (IFN-γ), tumor necrosis factor-α (TNF-α), and by activating NKT, natural killer (NK), and T cells (Fig. 1) [17].
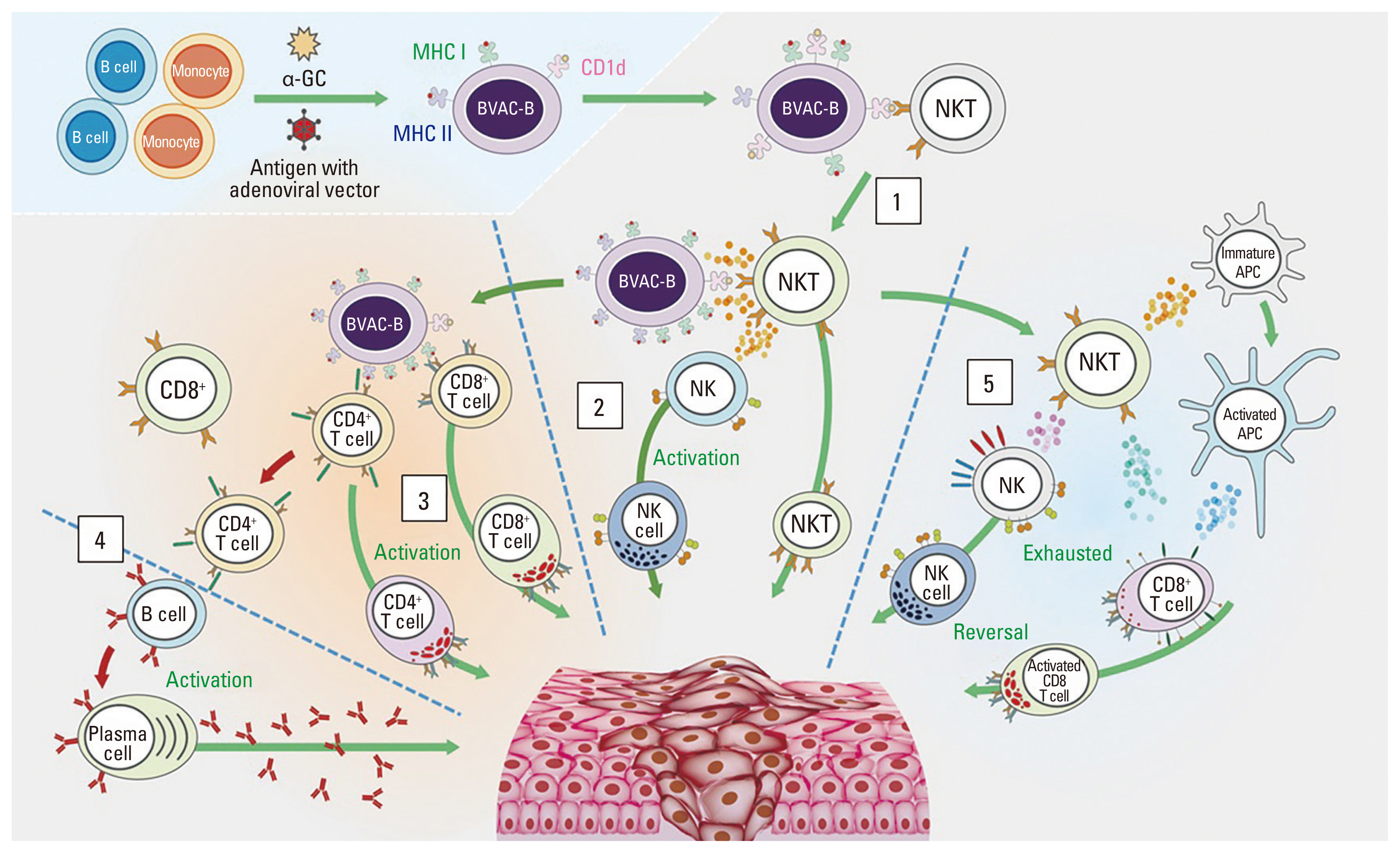
Mode of action of BVAC-B. After BVAC-B is injected into the human body, it interacts with and activates NKT cells (1). Activated NKT cells secrete cytokines such as IFN-γ and IL-4, which activate NK cells. Activated NK and NKT cells promote anti-tumor immune responses (2). BVAC-B also activates both antigen-specific CD4+ and CD8+ T cells. Cytokines secreted by activated NKT cells facilitate this process (3). CD8+ T cells differentiate into tumor-killing CTLs, which directly kill cancer cells. Activated CD4+ T cells activate CD8+ T cells, and B cells produce tumor Ag-specific antibodies (4). In addition, BVAC-B can induce the functional recovery of exhausted NK and CD8+ T cells through NKT cell activation (5). α-GC, α-galactosylceramide; APC, antigen presenting cell; CTL, cytotoxic T lymphocyte; IFN-γ, interferon gamma; IL-4, interleukin-4; MHC, major histocompatibility complex class; NK, natural killer; NKT, natural killer T cell.
We report a phase 1, first-in-human study to evaluate the safety and dose-limiting toxicity (DLT) of BVAC-B in treatment-refractory patients with HER2-positive gastric tumors. We also assessed the preliminary clinical efficacy and immune response after BVAC-B administration.
Materials and Methods
1. Patient eligibility
This open-label, first-in-human, phase 1 study of BVAC-B was conducted at Yonsei Cancer Center, Seoul, Korea. Patients with a histologically confirmed diagnosis of gastric adenocarcinoma with HER2-positive ≥ 1 based on immunohistochemistry (IHC) were selected for the trial. The inclusion criteria were as follows: (1) disease progression during standard treatment; (2) at least one measurable lesion defined according to Response Evaluation Criteria in Solid Tumor (RECIST 1.1); (3) age > 19 years; (4) an Eastern Cooperative Oncology Group performance status of 0 to 2; (5) adequate hematological (absolute neutrophil count ≥ 1,500/mm3 and platelet count ≥ 100,000/mm3), renal (serum creatinine ≤ 2.5 mg/dL or calculated creatinine clearance ≥ 30 mL/min), and liver function (serum bilirubin ≤ 1.5 upper limit of normal [ULN], transaminase ≤ 2.5 ULN); and (6) cardiac ejection fraction > 50%. Key exclusion criteria included the following: (1) treatment with corticosteroids, immunosuppressants, immune-modulating agents, granulocyte colony-stimulating factor, live vaccines, or any other investigational product within the previous 4 weeks and (2) history of heart failure, coronary artery disease, or myocardial infarction within 6 months prior to treatment initiation.
All participating patients provided informed consent. This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the institutional review board of our institution (IRB 4-2017-0606). This study is registered and is available from: ClinicalTrials.gov as NCT03425773 and was presented in part at the 2020 American Society of Clinical Oncology annual meeting [18].
2. Preparation of BVAC-B
Peripheral blood mononuclear cells (PBMCs) were collected from each patient by apheresis and transported to the Good Manufacturing Practice facility. Red blood cells were lysed using hypotonic buffer, and CD3+ T cells were depleted with magnetic cell separation (Miltenyi, Bergisch Gladbach, Germany). The remaining cells were transfected with recombinant HER2 and loaded with alpha-galactosylceramide. Subsequently, cells were harvested and frozen. Cells were placed in cryogenic vials and stored in nitrogen vapor before treatment.
3. Study design
BVAC-B was injected intravenously every 4 weeks for a total of four doses, and the total treatment duration was 18 weeks, including a follow-up period of 2 weeks after the last dose. The study was conducted in an accelerated titration dose escalation design, and one patient was initially assigned to each of the following dose groups: 2.5×107 cells/dose (low dose), 5.0×107 cells/dose (medium dose), and 1.0×108 cells/dose (high dose) [19]. Additional patients were recruited in the standard 3+3 design after evaluation of DLT within 2 weeks after the first injection. The National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI CTCAE ver. 4.03) were used to assess adverse events [20]. DLT was defined as > grade 3 cytokine release syndrome or grade 3 adverse events related to BVAC-B within 2 weeks after the first dose.
Tumor samples were tested with IHC (HercepTest, Dako, Glostrup, Denmark) and/or silver in-situ hybridization (SISH; HER2 SISH pharmDx, Dako) for HER2 2+ by IHC. Response evaluation with RECIST ver. 1.1 was assessed optionally at 4 weeks after the first injection and mandatorily after completion of treatment at 16 weeks. Anti-tumor activity was evaluated later than the standardized protocol of every 2 months because of the time required for an immune response to be elicited. For exploratory analysis, blood samples were collected prior to treatment, 24 hours after BVAC-B administration, every 2 weeks during treatment, and at the end of treatment.
The primary endpoint of this study was the safety and maximum tolerated dose (MTD) of BVAC-B therapy. Secondary endpoints included DLT, preliminary clinical efficacy, and BVAC-B–induced immune response. Endpoints for clinical efficacy included ORR, PFS, and OS. PFS was defined as the time from apheresis to disease progression or death from any cause. OS was defined as the time from apheresis to death from any cause. BVAC-B-induced immune responses included serum cytokine analysis for IFN-γ, TNF-α, interleukin 4 (IL-4), IL-6, and HER2-specific immune responses.
4. Immunological responses
1) Serum cytokine analysis
We collected and cryopreserved patients’ blood samples for IFN-γ, TNF-α, IL-4, and IL-6 analysis before and 24 hours after each BVAC-B infusion. Cytokines were measured using Milliplex (Millipore, Burlington, MA) and analyzed using a Luminex 200 microbead analyzer (Millipore), according to the manufacturer’s instructions.
2) Ex vivo IFN-γ ELISPOT
For ex vivo IFN-γ ELISPOT, after stimulation with a HER2-specific antibody, PBMCs from each patient were collected and cryopreserved every 2 weeks after the first infusion. Cryopreserved PBMCs were thawed and cultured with X-VIVO15 (Lonza, Köln, Germany) for more than 16 hours at 37°C and 5% carbon dioxide (CO2). PBMCs (2×105 cells per well) were subsequently stimulated with 5 μg/mL HER2-derived 15-mer peptides with overlapping seven amino acids for 48 hours. Anti-human CD3 antibody (OKT3, BioLegend, San Diego, CA) and medium served as positive and negative controls, respectively. After stimulation, spots indicating IFN-γ–secreting cells were monitored according to the manufacturer’s instructions (Cellular Technology, Ltd., Shaker Heights, OH). The IFN-γ spot ratio, relative to T-cell ELISPOT, was calculated using the following formula:
3) Detection of HER2-specific antibodies
For detection of HER2-specific antibodies, CT26/HER2 cells were opsonized with serially diluted plasma from patients and washed with Dulbecco’s phosphate-buffered saline (Thermo Fisher Scientific, Pittsburgh, PA). After opsonization, CT26/HER2 cells were stained with a PE-conjugated anti-human IgG Fc antibody and analyzed with flow cytometry.
5. Statistics
All patients who received at least one dose of BVAC-B were included in the analysis of the intent-to-treat population. Data were analyzed using the Statistical Package for the Social Sciences ver. 25 (IBM Corp., Armonk, NY) and GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA). The Kaplan-Meier method was used to analyze the median PFS and OS, and Student’s t test was performed to analyze differences in parameters before and after BVAC-B treatment. Safety and tolerability were analyzed using descriptive statistics.
Results
1. Patient characteristics
Between February 23, 2018, and April 23, 2019, a total of 0 patients were screened, and eight patients were eligible for enrollment in the study. The data cutoff date was February 5, 2020. One patient was enrolled in each vaccine dose group during the accelerated titration stage. Since no DLT was observed in any stage, five additional patients were enrolled in the high-dose cohort.
Patient characteristics are shown in Table 1. Four patients (50%) were men, and the median age was 51 years (range, 40 to 80 years). Six patients (74%) were 3+, one patient was 2+ (with an amplification index of 4.42, SISH), and one patient was 1+ by HER2 IHC. The median number of previous treatments was four (range, 1 to 8), and seven patients (87%) had three or more metastatic sites prior to treatment with BVAC-B.

Patient characteristics
2. Adverse events
No treatment-related adverse events (TRAEs) were observed in the low-dose cohort. Grades 1 and 2 TRAEs were observed only in patients treated with medium or high doses (Table 2). The most common adverse event was fever of grade 1 (G1; n=2, 25%) and grade 2 (G2; n=2, 25%), which was managed with supportive care. Other adverse events included G1 of (1) increase in aspartate aminotransferase (n=1, 13%), (2) alanine transaminase (n=1, 13%), (3) hypotension (n=1, 13%), and (4) cytokine release syndrome (n=1, 13%). No patient experienced grade 3 or 4 TRAEs or discontinued treatment owing to toxicity issues. The MTD was not reached, and 1×108 cells/dose (high dose) BVAC-B was tolerated in all additional six patients in the high-dose group.

Treatment-related adverse events
3. Clinical efficacy
All eight patients treated with at least one dose of BVAC-B were included in the analysis of clinical responses (Fig. 2). Patients in the low- and medium-dose cohorts underwent only one cycle of BVAC-B treatment owing to rapid progression. At the high dose, two patients underwent only one treatment cycle, three patients underwent two treatment cycles, and only one patient completed all four treatment cycles (Table 3). Five patients (one patient had progression of non-target lesion) discontinued treatment because of disease progression and three patients [S05(303), S07(305), and S10(306)] had stable disease (SD) according to RECIST at the first evaluation, as shown by the increase in tumor volumes, based on the waterfall plot (Fig. 2A). The three patients with SD had one or two prior chemotherapies and were confirmed to be HER2 3+ in the tumor tissues 5 months before BVAC-B treatment after first-line treatment. However, no patient showed partial or complete response. The swimmer plot shows the PFS for each participant (Fig. 2B). At the time of the cutoff for analysis, all patients died. Five patients (63%) underwent subsequent treatments. The median PFS and median OS were 1.17 months (95% confidence interval [CI], 0.15 to 2.18 months) and 4.1 months (95% CI, 0 to 7.7 months), respectively (Fig. 2C and D). A summary of the clinical and immunological responses is presented in Table 3.

Tumor response. Changes from baseline in the sum of the largest target lesion diameters per patient (A). Swimmer plot for PFS and subsequent treatments (B). Kaplan-Maier’s curve for PFS (C) and OS in eight patients (D). CI, confidence interval; OS, overall survival; PD, progression of disease; PFS, progression-free survival; SD, stable disease.

Clinical and immune responses
4. Immunological responses
1) Serum cytokine analysis
We evaluated changes in serum cytokine levels related to immune responses. Fig. 3 shows the serum cytokine levels measured before and 24 hours after BVAC-B administration during each cycle (Fig. 3A). IFN-γ, TNF-α, and IL-6 increased at 24 hours after each BVAC-B administration in all patients, except for the patient treated with a low dose (patient 101). Interestingly, patients treated with high dose BVAC-B [S06(304), S07(305), S10(306)] showed subsequent elevation of IFN-γ and TNF-α (Fig. 3B, C and E). However, IL-4 levels only slightly increased in three patients (Fig. 3D). In addition, the relative IL-6 level 24 hours after the 1st BVAC-B injection was significantly higher in patients with SD than in those with PD (p=0.009) (Fig. 3E).

Study flow chart and immune response results. Study flow chart for BVAC-B injection and blood sampling (A). Relative serum cytokine changes at each time point; IFN-γ (B), TNF-α (C), IL-4 (D), and IL-6 (E). Relative IFN-γ spot ratios by ELISPOT (F). Relative HER2-specific antibody at each time point (G). HER2, human epidermal growth factor receptor 2; IFN-γ, interferon gamma; IL-4, interleukin-4; IL-6, interleukin-6; PD, progression of disease; SD, stable disease; TNF-α, tumor necrosis factor-α; w, week. a)Statistically significant between patients with PD and SD.
2) Ex vivo IFN-γ ELISPOT
The IFN-γ spot ratio by T-cell ELISPOT assay was evaluated in all eight patients. The relative IFN-γ spot ratio at 2 weeks after BVAC-B treatment increased in S01(101) and S06(304), and at 4 weeks after BVAC-B treatment in S01(101) and S05(303) (Fig. 3F). However, the IFN-γ spot ratio did not increase in S02(201), S03(301), S04(302), S07(S305), and S10(306), and there was no difference between treatment responses (SD vs. PD) or BVAC-B dose levels (low, medium, and high dose).
3) HER2-specific antibodies
HER2-specific antibodies were detected in five patients: S01(101), S02(201), S03(S301), S04(S302), and S07(S305) (Fig. 3G). In addition, the relative HER2-specific antibody levels gradually increased in S01(101) and S02(201); however, the level of antibody was not higher in patients treated with high dose BVAC-B (vs. low or medium dose), nor in patients with SD compared to patients with PD.
Discussion
This study, which is the first human study examining BVAC-B monotherapy showed that BVAC-B had a safe toxicity profile with immunological activities in HER2-positive gastric cancer patients. However, clinical activities were limited and no adverse events or immunological responses were observed at low dose BVAC-B. No DLT was observed at any of the three doses, and all TRAEs were manageable; therefore, a high dose (1×108 cells) was considered as the recommended dose for further studies.
In the last two decades, three types of cancer immunotherapy, immune checkpoint inhibitors (ICIs), adoptive cell therapy (ACT), and anti-tumor vaccines, have shown promising results and have changed the new standard treatments against various cancers [21]. ACT, especially CAR T cell therapy, has shown prolonged OS for hematological malignancies [22]; however, it is not an effective treatment option for solid tumors, including HER2-positive cancers [23]. The lack of response to CAR T cells in solid tumors may be attributed to various reasons: (1) There is no ideal antigen that is specifically expressed in solid tumors; rather, tumor-associated antigens (TAAs) are also expressed at low levels in normal tissues; (2) existence of multiple barriers, including physical barriers such as cancer-associated fibroblasts and distorted vasculature at the tumor site, hinder the CAR T cells from reaching the tumor site, and (3) presence of a hostile TME, including hypoxic and glycolytic mechanisms, and suppressive immune cells that prevent proper function of CAR T cells [24].
Therapeutic anti-cancer vaccines have been developed as alternative treatment options for patients who do not respond to ICIs and CAR T cells. Several attempts have been made to target pathways of immune evasion using vaccines against gastric cancer [24]. One example is dendritic cells (DCs) pulsed with HER2/NEU-derived peptides administered to gastric cancer patients, which elicited immune responses in nine patients [24]. Among the patients, one patient achieved a partial response, and one patient had SD. Other DC vaccines have also been developed, although they have limited efficacy owing to the short lifespan of DCs [24]. Notably, OTSGC-A24, an anti-cancer vaccine targeting TSAs including FOXM1, DEPDC1, KIF20A, URLC10, and vascular epithelial growth factor receptor 1, showed cytotoxic T lymphocyte (CTL) activity in patients with advanced gastric cancer harboring the human leukocyte antigen (HLA)-A 24:02 haplotype [25]. A phase 2 study of OTSGC-A24 in combination with immunotherapeutic agents such as nivolumab and ipilimumab is ongoing (NCT03784040).
Previously, the disparity between preclinical endpoints and clinical response has been attributed to the use of surrogate endpoints, such as tumor necrosis and lymphocyte infiltration, instead of estimating directly the percentage of tumor shrinkage that accurately reflects clinical validation [26]. Several factors hinder the development of anti-cancer vaccines. One of the most important challenges is the identification of TSAs [26]. Comparing to TAAs are overexpressed protein in cancer cell and normal cells, TSAs are only expressed in cancer cells and not in normal cells or tissues [27]. Therefore, TSAs are ideal targets for cancer immunotherapy because the human immune system has not previously been exposed to TSAs and can recognize TSAs as non-self and trigger strong immune response. However, HER2/NEU-derived peptides are not TSAs, but TAAs, T and B cells might have immunologic tolerance with reactivity to self-antigens [27]. Therefore, TAAs targeted in anti-cancer vaccines should be (1) expressed only in cancer cells, (2) generally presented by cancer cells, (3) immunogenic, and (4) necessary for cancer cell survival. Furthermore, antigens should be recognized by immune cells in vivo to induce cellular immune responses. Anti-cancer vaccines must activate quiescent immune cells in the TME to achieve tumor shrinkage [26]. Especially, as several CTL eptiopics are restricted by specific HLA type, such as immunogenic HER2-derived peptides E75 (KIFGSLAFL) with high affinity for HLA-A2 and HE1 (RWGLLLALL) with HLA-A24 [28,29]. In the current study, we tested HLA typing in subject treated with high dose of BVAC-B (S1 Table). S07 and S10 subjects with HLA-A24 showed SD, although we could not conclude the association between HLA typing and BVAC-B efficacy because of the limitation of sample size and low efficacy, the subjects with HLA-A24 might have responsed more with BVAC-B. Furthermore, next studies should consider transformed BVAC-B with a strong binder sequence of HER2 in subjects with a specific HLA type for better immune response.
Compared to DC vaccines, BVAC-B has clinical usefulness. Manufacturing DC vaccines is time- and labor-consuming because the number of DCs is low among PBMCs. In contrast, B cells and monocytes are abundant within PBMCs; therefore, they are easily collected by apheresis. In addition, BVAC-C, which has a mode of action similar to that of BVAC-B, showed durable anti-tumor activity and immune response in HPV 16/18-positive cervical cancer patients [18]. BVAC-B activates NKT cells, which secrete IFN-γ and stimulate NK cells. Therefore, we assessed the level of plasma cytokines and PBMCs in each patient to evaluate innate and adaptive immune responses. Our exploratory analysis showed that serum IFN-γ and TNF-α, which are associated with NKT and NK cell–related inflammation, increased after BVAC-B treatment. In addition, we observed antigen-specific immune responses with increased levels of cytokines (IFN-γ, TNF-α, and IL-6) and anti–HER2/NEU specific antibodies in a subset of patients; especially, the level of IL-6 24 hours after BVAC-B injection was significantly increased in patients with SD than in those with PD. Despite the feasibility and tolerability of BVAC-B, no objective responses were observed because most patients [S01(101), S02(201), S04(302), and S06(304)] who were heavily treated (≥ 4 prior line of treatment) had a high disease burden (≥ 3 metastatic lesions) and underwent less than four BVAC-B cycles. In contrast, three patients [S05(303), S07(305), and S10(306)] who achieved SD underwent only one or two cycles of prior treatment. In general, tumor cell killing by the immune system is suppressed at high tumor burdens [26].
Considering the clinical efficacy of BVAC-B and BVAC-C, BVAC-B may have better clinical efficacy for HER2 strong positive (IHC 2+/fluorescence in situ hybridization+ or IHC 3+) gastric tumors, but it was not properly assessed owing to the lack of available tumor tissue on post-progression biopsy from prior treatment. The HER2 test was performed using tumor tissue when patients were initially diagnosed with advanced or metastatic gastric cancer. In fact, some studies reported approximately 30% loss of HER2 positivity on post-progression biopsy tissue after anti-HER2 therapy in gastric cancer patients [29]. Therefore, to test for the actual clinical efficacy of BVAC-B on HER2 positivity, it is necessary to re-test HER2 positivity using post-progression tissue before enrollment. In addition, three patients [S05(303), S07(305), and S10(306)], who achieved SD, were re-tested for HER2 status within 5 months before BVAC-B treatment, and all cases were confirmed as HER2 3+ after first-line treatment. Furthermore, in contrast to cytotoxic chemotherapeutic agents that kill cancer cells directly, immunotherapy, including anti-cancer vaccines, requires a longer time to elicit clinical responses because of the time required for immune cells to become activated [30]. A better understanding of the immunosuppressive environment is needed to overcome the limitations of therapeutic anti-cancer vaccines, including BVAC-B. Combination treatment of anti-cancer vaccines with immunotherapeutic or cytotoxic agents may help overcome the immunosuppressive tumor environment [24].
Major limitations in this study did not report the association between HER-specific peptide-MHC–specific immune response in subjects. Second, there was a lack of a sufficient number of patients to analyze both the efficacy and immune responses of BVAC-B. Most patients stopped treatment without completing all four cycles of BVAC-B owing to radiological and clinical progression. In addition, analysis of antigen-specific immune responses to BVAC-B was performed in only a subset of patients because of the early termination of the study, leading to a paucity of immune response data.
In conclusion, BVAC-B showed an acceptable toxicity profile in patients with HER2 IHC ≥ 1 positive acute gastric cancer. MTD of BVAC-B was not achieved, and the tolerability of high-dose BVAC-B provides a rationale for future combination treatments. To examine clinically relevant outcomes, further studies with earlier application of BVAC-B, combination treatments with other immunotherapeutic agents or modified BVAC-B with strong binder sequence of HER2 in subjects with a specific HLA type are warranted.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the institutional review board of our institution (IRB 4-2017-0606). All participating patients provided informed consent.
Author Contributions
Conceived and designed the analysis: Jung M, Kim HS, Kim HO, Kim S, Park M, Kim W, Choi KY, Oh T, Kang CY, Chung HC, Rha SY.
Collected the data: Jung M, Kim HS, Kwon WS, Rha SY.
Contributed data or analysis tools: Jung M, Lee JB, Kim HS, Kwon WS, Kim HO, Kim S, Park M, Kim W, Choi KY, Oh T, Kang CY, Chung HC, Rha SY.
Performed the analysis: Jung M, Lee JB, Rha SY.
Wrote the paper: Jung M, Lee JB, Rha SY.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgments
The results of this study were presented in part at the American Society of Clinical Oncology in 2020. We thank patients, caregivers, and family members who participated in the study. We also thank the members of Cellid, Inc., for their research on BVAC-B.