Functional Link between DNA Damage Responses and Transcriptional Regulation by ATM in Response to a Histone Deacetylase Inhibitor TSA
Article information
Abstract
Purpose
Mutations in the ATM (ataxia-telangiectasia mutated) gene, which encodes a 370 kd protein with a kinase catalytic domain, predisposes people to cancers, and these mutations are also linked to ataxia-telangiectasia (A-T). The histone acetylaion/deacetylation- dependent chromatin remodeling can activate the ATM kinase-mediated DNA damage signal pathway (in an accompanying work, Lee, 2007). This has led us to study whether this modification can impinge on the ATM-mediated DNA damage response via transcriptional modulation in order to understand the function of ATM in the regulation of gene transcription.
Materials and Methods
To identify the genes whose expression is regulated by ATM in response to histone deaceylase (HDAC) inhibition, we performed an analysis of oligonucleotide microarrays with using the appropriate cell lines, isogenic A-T (ATM-) and control (ATM+) cells, following treatment with a HDAC inhibitor TSA.
Results
Treatment with TSA reprograms the differential gene expression profile in response to HDAC inhibition in ATM- cells and ATM+ cells. We analyzed the genes that are regulated by TSA in the ATM-dependent manner, and we classified these genes into different functional categories, including those involved in cell cycle/DNA replication, DNA repair, apoptosis, growth/differentiation, cell- cell adhesion, signal transduction, metabolism and transcription.
Conclusions
We found that while some genes are regulated by TSA without regard to ATM, the patterns of gene regulation are differentially regulated in an ATM-dependent manner. Taken together, these finding indicate that ATM can regulate the transcription of genes that play critical roles in the molecular response to DNA damage, and this response is modulated through an altered HDAC inhibition-mediated gene expression.
INTRODUCTION
Ataxia-telangiectasia mutated (ATM) serine-threonine kinase globally regulates diverse cellular responses such as the genomic integrity, cell cycle checkpoints, DNA repair and the gene expression and apoptosis in response to genotoxic DNA damaging stresses (1,2). Accordingly, mutations of the ATM gene are directly linked to progressive and degenerative A-T, which is characterized by cerebellar degeneration, immunodeficiency, premature aging, rediosensitivity and a predisposition to cancers (1,2). These complex and diverse symptoms of A-T reflect the critical role of ATM (1,2).
ATM responds to DNA damage by activation of signal transduction through phosphorylation of a number of downstream substrates, including a downstream effector kinase Chk2, BRCA1/Rad51/BRCA2 and p53/MDM2 (1~4). Phosphorylation of these proteins plays a critical role in regulating their functions for the appropriate DNA damage responses (1~4). The role of ATM-mediated phosphorylation of the transcriptional activator p53 in the responses to DNA damage is well established (5). In addition, the transcription factors BRCA1 and CtIP mediate DNA damage responses via ATM-induced phosphorylations (6,7). It was recently reported that phosphorylations of Rb (8) and Che-1 (9) by the ATM/Chk2 pathway link transcriptional regulation to the DNA damage response. Although these evidences strongly suggest that ATM mediates DNA damage responses through transcriptional reprogramming after the DNA damage, the function of ATM in the regulation of gene transcription is not yet fully understood.
Histone deacetylases (HDACs) and acetyl transferases (HATs) are enzymes that catalyze the removal or addition of acetyl groups from lysine residues of histone tails by reversibly inducing hypo- and hyper-acetylation of histone, respectively, and this leads to chromatin remodeling (10). Alterations in the chromatin structure, including this post-translational modification of histone tails, allow the access of relevant proteins to the genome, and this is correlated to regulation of replication, the gene expression, DNA repair, the structure of the pericentirc heterochomatin and centromere, and the epigenetic integrity (10). Thus, chromatin remodeling by HDACs can regulate diverse cellular processes such as differentiation, replication, the cell cycle and the genomic integrity (10). The HATs and HDACs can modify various non-histone proteins, including p53, Ku 70 and AML, and thereby they regulate transcription, DNA repair and the cell cycle checkpoints (11~13). Furthermore, exposure of cells to chromatin-modifying drugs such as a HDAC inhibitor TSA induces the ATM-mediated DNA signal pathway (in an accompanying work, 14) and rapid, diffuse phosphorylation of the ATM protein (15); this suggests that ATM activation may result from changes in the chromatin structure. In addition, highly decondensed chromatin has been observed in A-T (16), supporting that ATM regulates the chromatin structure and transcription. Taken together, these observations suggest that histone acetylation functionally interplays with the ATM-mediated DNA damage responses in many ways. However, the function of ATM in the histone acetylation-mediated gene expression has not yet been investigated.
Here, we investigated the function of ATM in the regulation of gene transcription in response to HDAC inhibition. Thus, we examined the differential gene expression profiles of the ATM-regulated genes in isogenic A-T cells (ATM-) and in control cells (ATM+) following treatment with TSA. We identified the HDAC inhibition-regulated genes that are under the control of ATM by comparing the regulated genes in the ATM+ cells with those in the ATM- cells following treatment with TSA. These genes we identified are functionally diverse, which is consistent with the pleiotropic A-T phenotype. Our results have implications that ATM can function in diverse DNA damage responses through its ability to transduce stress signals (in an accompanying work), as well as reprogramming the gene expression profiles. Taken together, our findings indicate that the activation of ATM can work in the regulation of transcription to display the stress response phenotype in response to TSA.
MATERIALS AND METHODS
1) Cell culture and treatment
AT22IJE pEBS7 (ATM-) and AT22IJE pEBS7-YZ5 (ATM+) cells (gifts from Y. Shiloh) were cultured in DMEM supplemented with 100 g/ml hygromycin B and 15% fetal bovine serum (FBS, Hyclone, Logan, UT). The HCT 116 cells were cultured in DMEM supplemented with 10% FBS. The ATM- and ATM+ cells were treated with 0.33 M TSA for 24h before being harvested.
2) Oligonucleotide microarray analysis
The isolated total RNA samples were processed as recommended by Affymetrix (Affymetrix GeneChip Expression Analysis Technical Manual, Affymetrix, Inc.). In brief, double-stranded cDNA was synthesized from the poly (A)+ mRNA present in the isolated total RNA (30 g of total RNA starting material in each sample reaction). A portion of the resulting double-stranded cDNA was used as a template to generate biotin-tagged cRNA from an in vitro transcription reaction (IVT). 20 g of the resulting biotin-tagged cRNA was fragmented to strands of 35 to 200 bases in length, with following the prescribed protocols. Subsequently, 15 g of this fragmented target cRNA was hybridized at 45℃ with rotation for 16 h (Affymetrix GeneChip Hybridization Oven 640) to probe the sets that are present on an Affymetrix HG-U133A array. The GeneChip arrays were washed and then stained (streptavidin-phycoerythrin) on an Affymetrix Fluidics Station 400; this was followed by scanning on an Agilent GeneArray scanner (Agilent Technologies, Palo Alto, CA). The results were quantified and analyzed using MicroArray Suite 5.0 software (Affymetrix, Inc.). The data analysis was done as described in a previous report (2).
3) RNA extraction and RT-PCR
The total cellular RNA was isolated from the cultured cells using the TRIzol reagent (Life Technologies, Inc., Grand Island, NY) according to the manufacturer's instructions. The primer sequences are as follows: GAPDH (forward 5' GTCAACGGATTTGGTCTG TTT-3', reverse 5'-AGTCTTCTGGGTGGCAGTGAT-3'); BAX (forward 5'-GGGGC GAACTGGACAGTAA-3', reverse 5'-CAGTTGAAGTTGCCGTCAGA-3') (17); ERCC3 (forward 5'-CCAGGAAGCGGCACTATGAGG-3', reverse 5'-GGTCGTCCT TAGCGGCATTT-3') (18); ERBB2 (forward 5'-GAAGGTGAAGGTGCTTGGATCT-3', reverse 5'-TAGCTCCCCTTGGCAATCTGC-3'); CDKN1C (forward 5'-CGATCAAA AGCTGTCCGGGCCTC-3', reverse 5'-CCGCCGGTTGCTGCTACATGAACG-3') (19); CCND1 (forward 5'-CCTCTTGTGCCACAGATG-3', reverse 5'-GATGTCCAC GCCCGCAC-3'); CCND2 (forward 5'-TCATGACTTCATTGAGCA-3', reverse 5'-CA CTTCCTCATCCTGCTG-3') (20); ATM-p1 (exon 7-10) (forward 5'-ATGCAGGAAA TCAGTAGTTT-3', reverse 5'-AAAGCAAAGTGGGAAGAATT-3') (21); ATM-p2 (exon 10-13) (forward 5'-ATTCTTCCCACTTTGCTTT-3', reverse 5'-ATAAATCTGA CTTTTGTGAG-3') (21), and MCL1 (forward 5'-CTTCCGTAATTAGGAACCTG-3', reverse 5'-CTTGCATATAATGAAGTGAA-3') (22). All the PCR products were analyzed in 1.2% agarose gels.
4) Transient transfection and chemical treatment
The HCT 116 cells were transfected with the Flag-tagged wild type (Flag-ATM-WT) and the kinase-dead ATM (Flag-ATM-KD) expression vectors (16) with using the Effectene transfection kit (Qiagen). One day after transfection, the cells were incubated for another 24 h with 0.33 M TSA before being harvested. Trichostatin A (TSA) and wortmannin were purchased from Sigma Chemical Co.
RESULTS
1) Oligonucleotide microarray analysis of the expression profile of the ATM-regulated genes following HDAC inhibition
To examine the expression profiles of the ATM-regulated genes in response to HDAC inhibition, we performed microarray analysis of the ATM+ and ATM- cells that were treated with or without 0.33 M of the HDAC inhibitor TSA for 24 h. This dose and time point were previously determined as being optimal for the DNA damage signals (14).
We identified the TSA-responsive genes (using a cut-off ratio of 2) by comparing the gene expressions before and after TSA-treatment in the ATM+ and ATM- cells (Fig. 1). In the ATM+ cells, HDAC inhibition by TSA induced 1021 genes and this repressed 1302 genes (Fig. 1A). In the ATM- cells, 534 genes were induced and 477 genes were repressed by TSA treatment (Fig. 1B). The total number of TSA-responsive genes in the ATM+ cells was 2-fold that in the ATM- cells, suggesting that cells may be able to dynamically respond to HDAC inhibition in the presence of ATM.
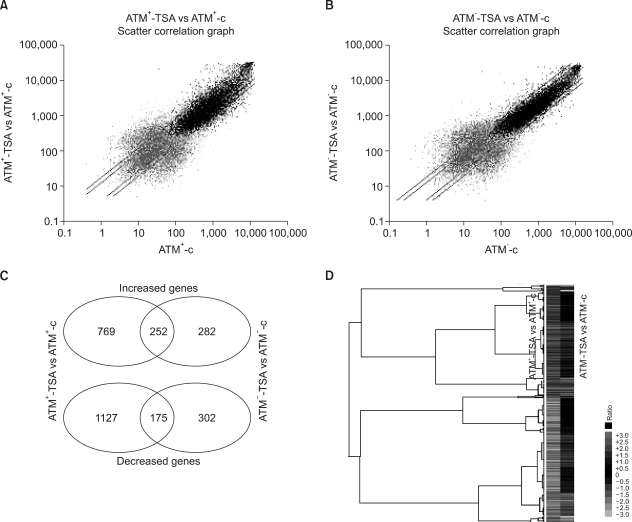
Comparison of the gene expression profiles between the ATM- and ATM+ cells in response to TSA, as was derived from the oligonucleotide microarray analysis. (A) The signal intensities from the TSA-treated ATM+ cells (y-axis) relative to those from the untreated ATM+ cells (x-axis) were analyzed using MicroArray Suite 5.0 software. The log-transformed data is shown; the guideline shows the 2-fold signal ratio. (B) The signal intensities in the ATM- cells. The ATM-dependent and ATM-independent TSA-responsive genes were distinguished from the Venn diagrams (C), and the genes that were significantly enhanced or reduced in the ATM+ cells, as compared to that in the ATM- cells, in response to TSA were visualized by Treeview software (D). The expressions of more genes are regulated in the ATM+ cells, as compared to the ATM- cells, in response to HDAC inhibition.
Among the TSA-responsive genes, 252 and 175 genes were up-regulated or down-regulated by HDAC inhibition, respectively, in both the ATM+ and ATM- cells (Fig. 1C), suggesting that their expressions may be regulated by HDAC inhibition in an ATM-independent fashion. In contrast, 769 and 1127 genes showed increased or decreased signals, respectively, in only the ATM+ cells, indicating that these genes were regulated by TSA-treatment in an ATM-dependent manner. Additionally, 282 and 302 genes were up-regulated and down-regulated, respectively, in the ATM- cells, indicating that these genes were regulated by TSA treatment in the absence of ATM. Furthermore, when the ratios of the gene intensities in the TSA-treated ATM+ cells versus those of the non-treated ATM+ cells were compared with the ratios of the intensities in the TSA-treated ATM- cells versus those in the untreated ATM- cells, the results were consistent with our comparison between the signals before and after TSA treatment in each type of ATM cell (Fig. 1D).
The identified TSA-responsive genes mediated by ATM are shown in Table 1. These included genes related to chromatin remodeling, such as methyltransferases (GAMT) and histone family members (H2AFX, HIST3H2A, etc) (up-regulated), and methyltransferases (RNMT, NNMT, HRMT1L2, and CARM1) and a regulator of chromatin (SMARCC1) (down-regulated). In addition, diverse transcription-associated genes (RUNX1, JUND, NFκB2, TCF, RXR and CREB1) were regulated by TSA in an ATM-dependent manner. These included many known apoptosis-related genes such as TRADD, MCL1, NFκB2, Gadd45A, BAX, BCL2L, GRIM19 and TRAIL receptor 2 (TNFRSF10B), as well as a number of cell cycle/DNA replication/DNA repair-associated genes (CDKN1C, CCND2, CDK2, CDK4, CDC2, ERCC3, MCM7, aurora B, SKP2, NEK2, BUB3 and CENPF). Of these, some were also found in the DNA damage- responsive group (e.g. MCL1, JUND, SKP2 and TRAIL receptor 2), implying that these genes may form the basis of the complex stress response system.
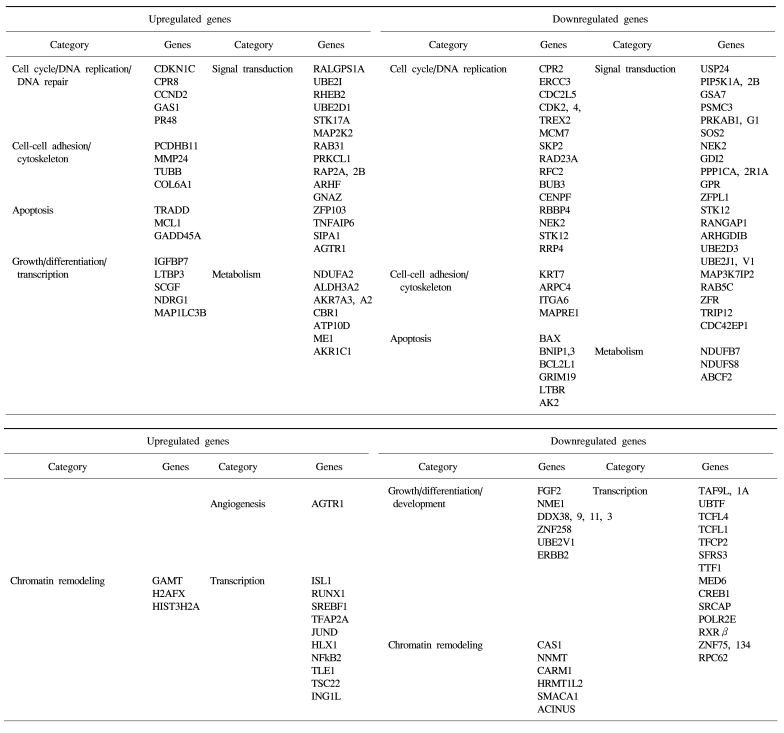
Classification of the ATM-regulated genes in response to TSA
2) Validation of the oligonucleotide microarray data using RT-PCR analysis
To validate these microarray results, we performed reverse transcription-polymerase chain reaction analyses (Fig. 2). To confirm the microarray data in regards to the TSA-responsive ATM target genes, we randomly selected 5 of these genes for further testing. Up-regulation of CDKN1C (cyclin-dependent kinase inhibitor 1C, p57, Kip2) and down-regulation of ERCC3 (excision repair cross-complementing rodent repair deficiency, complementation group 3), BAX, CCND1 (cyclin D1) and ERBB2 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog) was confirmed by our RT-PCR analysis (Fig. 2A).

The oligonucleotide microarray results were validated by RT-PCR analysis of the expression of selected genes in the ATM- cells, the ATM+ cells and the HCT116 cells. (A) The expressions of the TSA responsive genes were evaluated by RT-PCR as being representative of the ATM-regulated genes. The TSA-induced increase in the level of CDKN1C mRNA was reduced in the absence of ATM. The TSA-induced reduction of the expressions of ERCC3, BAX, CND1 and ERBB2 mRNAs was observed in the ATM+ cells, but not the ATM- cells. (B) The effect of wortmannin on the expression of the ATM-regulated genes in response to TSA. Inhibition of ATM kinase activity by wortmannin attenuated the TSA-induced transcriptional effects on upregulation of the CDKN1C and CCND2 genes. (C) The ATM-expressing constructs were transiently transfected into HCT116 cells, and the expression levels of ATM-WT or ATM-KD were checked by RT-PCR with using the primer sets ATM-p1 (exon 7~10) and ATM-p2 (exon 10~13). (D) Monitoring of the ATM pathway activation after etoposide treatment by measuring the Chk2 phosphorylation in the HCT116 cells expressing ATM-WT or ATM-KD. In response to etoposide, ATM-WT phosphorylated Chk2 and this resulted in the shifted mobility of the phosphorylated Chk2 proteins, compared with that of nonphosphorylated Chk2. (E) Evaluation of the mRNA expression of the genes regulated by TSA in the HCT116 cells expressing ATM-WT or ATM-KD. The expressions of BAX and CCND1 were reduced in response to TSA in the cells expressing ATM-WT, and that of CDKN1C increased in the cells expressing ATM-WT. TSA-induced downregulation of BAX and CCND1 and upregulation of CDKN1C was rarely detected in the cells expressing ATM-KD. GAPDH mRNA was amplified as an internal control.
3) ATM kinase activity is essential for ATM-mediated transcriptional modulation in response to HDAC inhibition
We next studied whether ATM kinase activity is necessary for ATM-mediated transcriptional regulation following HDAC inhibition. To this end, we tested the effect of the PI3K inhibitor wortmannin on the ATM-mediated gene expression changes following HDAC inhibition. Treatment with 10 M wortmannin abrogated the TSA-induced up-regulation of CDKN1C and CCND2 in the ATM+ cells (Fig. 2B). Additionally, the ERBB2 and ERCC3 genes were also regulated by wortmannin (data not shown). Taken together, these data suggest that ATM kinase activity is required for ATM-mediated TSA-induced transcriptional modulation.
To determine whether the expression of the ATM target genes in response to HDAC inhibition was mediated through ATM, we investigated the effect of exogenous ATM on the expression of these genes (Fig. 2C). HCT116 cells are normally unable to activate Chk2 following DNA damage (etoposide) (Fig. 2D). Using this system, we then studied the expression of the ATM target genes after treatment with TSA (Fig. 2E). As shown in Fig. 2E, the TSA-responsive genes BAX, CCND1 and CDKN1C were regulated in the ATM-WT-expressing cells in a manner similar to that in the ATM+ cells. Taken together, these results demonstrate that transcriptional regulation of the ATM target genes is mediated through ATM in response to TSA.
To determine whether ATM kinase activity is required for the ATM-mediated target gene expression, we transiently transfected a kinase deficient ATM (ATM-KD) into HCT-116 cells and we examined the expression of the ATM target genes after treatment with TSA (Fig. 2E). As shown in Fig. 2E, the TSA responsive genes BAX, CCND1 and CDKN1C were not significantly regulated in the ATM-KD-expressing cells, whereas they were appropriately regulated in the cells expressing ATM-WT. These results suggest that ATM-mediated transcriptional regulation of these genes in response to TSA is dependent on its kinase activity. Together, these data indicate that activation of the ATM signaling pathway is important for the transcriptional modulation mediated through ATM.
4) The transcriptional regulation of MCL1, an ATM target gene, in response to HDAC inhibition
To further study new target genes that are involved in ATM-regulated transcriptional modulation, we examined the expression profiles of the ATM-regulated genes in the ATM+ cells treated with TSA, and we compared these to the corresponding profiles in the ATM- cells treated with TSA (Fig. 1). We found that the MCL1 (myeloid cell leukemia sequence 1, BCL2-related) gene was up-regulated 2.23-fold in an ATM-dependent manner by TSA (Table 1, Fig. 1). MCL1 is a known anti-apoptotic gene and it is an important factor contributing to chemoresistance (23). Thus, we next tested whether and how MCL1 was involved in the transcriptional regulation of ATM+ cells by monitoring the MCL1 expression levels by performing RT-PCR (Fig. 3).

Effects of TSA on the expression of MCL1 mRNA. Time course of the effect of TSA on the expression of MCL1 mRNA in the 293T cells. The TSA induced-upregulation of MCL1 mRNA fluctuated during the 1~24 h time course. GAPDH mRNA was amplified as an internal control.
We used the 293T cell line to monitor the time course and dose range of the TSA-induced increase in the MCL1 mRNA. As shown in Fig. 3, the increase in MCL1 occurred rapidly at 1h after treatment with 1 M TSA and the increase was transient, and it returned to near baseline in 8 h. Yet it once more increased within 12~24 h, and the increase reached a peak within approximately 24 h after 1 M TSA treatment. Also, the MCL1 mRNA level was significantly increased by 1 M TSA after 1 h, 12 h and 24 h treatment more than that by any other dose, whereas the MCL1 mRNA level was not increased by 1 M TSA after 8 h treatment. Although the dose and time point of the increased MCL1 expression were exactly coincident with those of the TSA-induced DNA damage response, the MCL1 expression was increased by TSA in the DNA damage-induced time point and dose range. The increase of the MCL1 mRNA in response to TSA is reminiscent of the increase seen with irradiation (24).
DISCUSSION
When DNA is damaged, signal transduction pathways are activated to induce a variety of important cellular responses. The ATM-mediated signal transduction pathway is a prime element for the responses to DNA damage. Modulations in the expression of genes act as sequential intermediaries throughout the process, triggering the signaling events and final phenotypic responses that are displayed by downstream effectors. As such, the ATM pathway can also be considered to be an intermediate cellular response to DNA damage via transcriptional modulation. However, although ATM is known to play key pleiotropic roles in intracellular signaling and the responses to DNA damage, the precise functional interplay among the roles of ATM in transcriptional modulation and the responses to cellular DNA damage has not yet been elucidated. In this study, we explored the role of ATM in transcriptional reprogramming in response to DNA damage, and we did so by cataloging the ATM-regulated genes and analyzing the gene expression patterns regulated by different types of stresses mediated through ATM. We found that impaired ATM function altered the transcriptional reprogramming in response to DNA damage. Furthermore, we found that several of the identified ATM target genes acted in growth regulation, which is one of the major cellular responses to various stresses.
The ATM-p53 pathway is a well-established paradigm for transcription-mediated DNA damage responses, and it plays multiple roles in cell cycle checkpoints, genomic integrity, apoptosis, proliferation and senescence. When cells encounter environmental or cellular stresses, changes in transcriptional regulation act as an important adaptive mechanism, leading to stress-responsive phenotypes. Several transcription factors are known to be responsive to DNA damaging stresses, including p53, NF-B, and the Sp1-related retinoblastoma control proteins (RCPs) (25). These previous observations underscore the role of ATM-p53 pathway mediated transcription in the responses to DNA damage, and they suggest that other ATM-transcription factor pathways may be involved.
Because of the important roles played by ATM in DNA damage responses (13~15), we studied the expression profiles of the ATM-responsive genes following HDAC inhibition in an effort to understand the functional link between HDAC activity and the ATM-mediated DNA damage responses. Concomitant to the markedly decreased expression of cyclin D1 in the ATM+ cells following HDAC inhibition, these cells showed growth arrest and increased apoptosis when compared to that in the parental ATM- cells (data not shown). Interestingly, HDAC inhibition also induced transcriptional repression of the ERBB2 gene and transcriptional activation of the TRADD and Gadd45 genes, as was shown by our microarray data. The originally radioresistant ATM+ cells showed these alterations of expression following HDAC inhibition, as was manifested by radiation sensitivity (repression of ERBB2) and apoptosis (induction of TRADD and GADD45). Because the molecular responses to DNA damage can be modulated through altered HDAC inhibition-mediated gene expression, we further questioned whether co-treatment of the radioresistant ATM+ cells with TSA and DNA damaging agents (either etoposide or cisplatin) might enhance the sensitivity to radiation or DNA damaging agents. Indeed, we found that the responsiveness to (radiation or) DNA damage was induced in the TSA-treated ATM+ cells, implying that the cellular responses to stresses are determined by the gene expression and thus, they can be regulated by modulation of the gene expression.
MCL1 can promote cell viability, although the effects of MCL1 may be more short-term than the effects of BCL2, and MCL1 is turn over more rapidly (24). Therefore, MCL1 may act as a short-term regulator in a labile mechanism for protection of viability. Surprisingly, our data showed fluctuations of the anti-apoptotic MCL1 expression. At the onset of the DNA damage response, the ATM-dependent loss of MCL1 may play a role in initiating the apoptotic pathway and this results in induction of apoptosis. However, its ATM-dependent restoration and the induction of MCL1 that occur as the intermediate events to the late DNA damage response may play a role in rescuing damaged cells and conferring survival, possibly leading to chemo-resistance or radio-resistance, and tumor recurrence. As such, these paradoxical functions of ATM in the transcriptional oscillatory reprogramming partially account for its roles in tumor suppression and therapeutic resistance.
Additionally, we analyzed the expression patterns of the previously identified ATM-mediated genes in the absence of stress (2) in the TSA-treated ATM+ and ATM- cells to see whether their expressions were altered by stresses in an ATM-dependent or independent fashion. The expression of most of the ATM-mediated stress-unrelated genes (2) remained unchanged in the ATM+ and ATM- cells in the presence of TSA. A total number of ATM-mediated genes in response to stress was bigger than that in the absence of stress (2). Furthermore, the absolute differences in the expression levels of the ATM-mediated stress-related genes increased or decreased dramatically in response to stress, suggesting that in terms of regulation of the gene expression, ATM functions more actively in the presence of stress. Taken together, these observations indicate that in the presence of stress, ATM may regulate a larger gene set than that in the absence of stress.
The A-T phenotype and our results imply that the stress response phenotype is determined by the gene expression and so this can be inferred by analysis of the stress-mediated genes in a given genetic background. The ATM protein can function within diverse DNA damage responses through its ability to transduce stress signals as well as reprogramming the gene expression profiles.
CONCLUSIONS
As demonstrated here, the altered gene expression profile directed by the ATM-mediated target genes can shape the cellular phenotypes in response to a variety of stresses. Thus, our study adds to our understanding of the transcription-mediated role of ATM. In addition, this work may provide valuable molecular therapeutic information for improving the clinical applications and courses of chemotherapy and radiotherapy for cancers, as well as aiding future attempts to treat the progression of A-T and similar DNA double strand breakage syndromes.
ACKNOWLEDGEMENTS
We thank the members of Lee's laboratory for their helpful comments on this study.
Notes
This work was supported by a research grant from Ajou University.