Dexamethasone Inhibits TRAIL- and Anti-cancer Drugs-induced Cell Death in A549 Cells through Inducing NF-κB-independent cIAP2 Expression
Article information
Abstract
Purpose
We have examined that dexamethasone inhibits apoptotic cell death of A549 lung epithelial cells through TRAIL and anti-cancer drugs. The purpose of the study is to determine the roles of GR, cIAP and NF-κB in this mechanism.
Materials and Methods
In the A549 lung epithelial cell line, TRAIL, taxol, doxorubicine & gemcitabine were used to investigate cell toxicity. Cells were pretreated 12 hours in advance with dexamethasone. RU486 was pretreated 30 minutes before dexamethasone. Crystal violet assay was used for cell toxicity tests. Apoptosis assay was performed by taking morphologic surveys with fluorescent microscopy after double staining with Hoechst 33342 & propium iodide. RT-PCR was used to investigate the gene expression of cIAP1 & cIAP2 by dexamethasone. Ad-IκB α-SR transduction study was used for the role of NF-κB.
Results
TRAIL and anti-cancer drug-induced apoptosis was partially suppressed in A549 cells pretreated with dexamethasone. The inhibitory effect on cell death disappeared in A549 cells pretreated with RU486. Using RT-PCR, changes of cIAP1 and cIAP2 genes manifestation in A549 cells subsequent to pretreatment with dexamethasone were examined. The results showed an increase in cIAP2 expression during a course of time which was suppressed by RU486 pretreatment. Induction of cIAP2 expression changes by dexamethasone was uniquely observed despite the blockade of NF-κB by Ad-IκBα-SR transduction.
Conclusions
These results suggest that dexamethasone inhibits TRAIL- and anti-cancer drug-induced apoptosis in A549 cells by inducing cIAP2 gene expression through a GR-mediated, NF-κB-independent pathway.
INTRODUCTION
Glucocorticoids are essential for survival and are involved in a variety of physiological processes including regulation of metabolism, immunity, and apoptosis (1). Glucocorticoids are widely used for their anti-inflammatory and immunosuppressive effects to a variety of diseases. Despite its wide use and effectiveness, the mechanism of its action became clearly known only a few years ago. However, it was recently discovered that steroid hormones affect the genetic transcription of cytokines such as TNF-α, IL-1β, IL-2, IL-3, IL-6, IL-11, and GM-CSF, chemokines such as IL-8, RANTES, MCP-1, MCP-3, MCP-4, MIP-1α, and eotaxin, as well as adhesion molecules (2,3).
The biologic action of glucocorticoids is mediated through the activation of intracellular glucocorticoid receptors (GR), ligand-activated transcription factors. Glucocorticoids pass freely through the cell membrane and enter the cytoplasm where they combine with inactive GR, utilizing a molecular chaperone, which is composed of two molecules: 90 kDa heat shock protein (hsp90) and 59 kDa immunophilin protein (3). When activated, the receptor translocates into the nucleus, dimerizes and binds to specific DNA sequences known as glucocorticoid response elements (GRE), and positively or negatively regulates transcription of target genes (2,4). It is also known that GR combines directly with inflammation-related transcription factors such as NF-κB, AP-1, CREB, OTF-1, and STAT5 and suppresses the effect of these transcription factors (4). Among these transcription factors, NF-κB has received the most attention due to its anti-inflammatory properties. The reaction of GR to glucocorticoids is affected by many factors in the surrounding environment, so it determines not just the concentration of GR, but also the interaction of GR with GRE, and with other transcription factors, such as AP-1 and NF-κB (2,5). That is, the enhancement and suppression of gene expression of GR is determined by specific characteristics of the cell or gene and by signaling stimulation or inhibition. The expression of GR is different in a variety of tissues and cell types, even the same types of tissue have minute differences between individual cells. The distribution and concentration of GR within cells and tissues affects the influence of glucocorticoids. GR is of the leucine zipper type of configuration, making it available for interaction with a variety of transcription factors. It is believed that this is an important mechanism which leads to its anti-inflammatory effect.
Glucocorticoids are known to have bivalent action in the aspect of cell death. Glucocorticoids are potent inducers of apoptotis in T-cells and monocytes, activating immunosuppressive and anti-inflammatory effects. Generally, apoptosis by T-cells is caused by activation from glucocorticoids binding to the T-cell receptor (TCR), causing a reaction with FasL. Binding to the Fas receptor then activates caspase 8 (6). Although the mechanism by which glucocorticoids cause apoptosis in T-cells is unclear, the activation of caspase 9 is known to induce apoptosis. This induction is known to be inhibited by Bcl-2 overexpression (7). However, T-cell apoptosis is suppressed if TCR and GR are activated simultaneously (8). Glucocorticoids prevent the induction of apoptosis through the induction of Bcl-2 and Bcl-XL in liver cells and glioma cells. Glucocorticoids also prevent induction of apoptosis due to taxol in human leukemic HL-60 cells and due to TNF-α in osteoblasts (9). In other words, even though glucocorticoids induce apoptosis in immunologic cells, they have a variety of different responses to apoptosis stimuli in different types of cells. However, the precise reason that glucocorticoids do or do not cause induction of apoptosis in various situations and cells is not clear.
An inhibitor of the apoptosis protein (IAP) family has been identified in baculoviruses. This inhibitor could inhibit the apoptotic response of insect cells to viral infection. In humans, there are six inhibitors of apoptosis proteins (IAPs) (XIAP/hILP, cIAP/HIAP2, cIAP2/HIAP1, NIAP, Survivin and BRUCE) currently known (10). Although the exact mechanism of the anti-apoptotic effect is not completely understood, reports have shown that the Baculovirus IAP Repeat (BIR) and RING domain both have roles, the BIR alone has an especially suppressive effect on apoptosis (11). A caspase recruit domain of cIAP1 and cIAP2 exists between the BIR and Ring domains, directly blocking caspase to inhibit apoptosis. Known as a human inhibitor of apoptosis 1 (HIAP1) or mammalian IAP homolog c (MIHC), cIAP2 was initially cloned as a protein that associates with the TNF receptor due to interaction with TNF receptor-associated factor (TRAF) 1 and 2 (12). The inhibitory cell death effect of cIAP2 is mediated by the TNF signal. NF-κB is activated to suppress the cell death which is mediated by TNF. This mechanism acts to directly bind caspase 3, 7, and 9 to cIAP2 and suppress caspases (10). In this way, the IAPs are believed to be involved in cancer development by the prevention of apoptosis induction through the suppression of caspases and control of TNF, Fas receptor, etc.
The expression of cIAP2 is enhanced by IL-1 and lipopolysaccharide in the vascular endothelium by CD40 stimulation in the B-cell. It has been determined that it is related to NF-κB stimulation (13). Presently, the most important transcription factor in cIAP expression is an NF-κB. In actuality, the cIAP gene promoter region has two NF-κB binding regions (14). That is, NF-κB dependent cIAP2 expression by TNF-α stimulation prevents the induction of apoptosis by that mechanism, as mentioned above, in which cIAP2 can bind to and directly inhibit caspase 3, 7, and 9, as well as suppress apoptosis. Also, cIAP2 stimulates NF-κB activation. In a similar way, the function of cIAP is well known, but the method of the regulation of expression is not well known. Until recently, it was believed that cIAP2 expression was solely regulated by NF-κB, but it was recently reported that glucocorticoids are also involved in cIAP2 expression (15).
We investigated the effect of glucocorticoids on TRAIL- and anticancer drug-induced cell death in A549 cells. Here, we show that dexamethasone inhibits the apoptotic death of A549 cells by TRAIL and anticancer drugs. Furthermore, we investigated the roles of GR, cIAP, and NF-κB in this mechanism. On the basis of our results, we conclude that dexamethasone inhibits TRAIL- and anticancer drug-induced apoptosis by cIAP2 expression through a GR-dependent, but NF-κB independent pathway in A549 cells.
MATERIALS AND METHODS
1) Cell and reagents
A549 cells were purchased from ATCC. Cells were cultured in appropriate media (RPMI 1640 in A549) with 10% fetal bovine serum supplemented with 2mM L-glutamine, 100 U/mlpenicillin and 100 U/ml streptomycin and cultured at 37℃, 5% CO2 incubator. Dexamethasone, RU486 and doxorubicine were purchased from Sigma (St. Louis, MO). TRAIL was purchased from Alexis Biochemicals (San Diego, CA), gemcitabine was obtained from Lilly (Indianapolis, IN), and taxol was obtained from BMS (Princeton, NJ).
2) Cell death assay
Cell viability was measured by a crystal violet staining assay. Cells were briefly treated with TRAIL, anti-cancer drugs and/or dexamethasone in a 24-well plate and were harvested at the indicated times followed by the addition of 500µl of crystal violet staining solution (0.5% crystal violet from 30% ethanol and 3% formaldehyde solution) for 15 minutes. The crystal violet staining solution was removed and cells were rinsed with distilled water four times each, then stored at room temperature for 30 minutes to dry. 500µl of 1% SDS solution was then added and 100µl was placed into 96-well plates. The absorbance at 590 nM was measured by an ELISA plate reader. Cell death was confirmed as apoptotic by taking morphologic surveys with a fluorescent microscope after double staing Hoechst 33342 and propium iodide.
3) Reverse-transcriptase polymerase chain reaction (RT-PCR)
To evaluate dexamethasone-inducing cIAP2 mRNA transcription, RT-PCR was used. A549 cells were cultured in a 60 mm dish until 80~90% confluency was achieved. A549 cells were treated with dexamethasone 1µM and culture medium was removed at 0, 1 hr, 2 hr, 4 hr after dexamethasone treatment and then Trizol (Gibco BRL, Gaithersburg, MD) 1 ml was added. A549 cells were cultured for 5 minutes at room temperature in an Eppendorf tube, then 200µl of chloroform was added and vortexed. After 2~3 minutes, it was centrifuged at 4℃ at 13,000 rpm. The RNA-inclusive solution was collected carefully and isopropyl alcohol 500µl was added. The solution was stored in ice for 10 minutes. It was then centrifuged at 13,000 rpm and the supernatant was removed, so isolating RNA. The isolated RNA concentration was measured using a photospectrometer (OD 260/280 nm, Beckman, CA). 2µg of RNA was added to oligo and a binding reaction was initiated at 65℃ for 5 minutes. First strand buffer, 0.1 M DTT, 25 mM dNTP, and RNAase inhibitor were added and a reaction was initiated at 42℃ for 2 minutes, then reverse transcriptase (RT) was added again and a reaction was initiated at 42℃ for 1 hr and cDNA was subsequently collected. cDNA was amplified in 10x PCR buffer, 2.5 nM dNTP, Taq DNA polymerase (TaKaRa TaqTM, TaKaRa, Japan). The PCRs were conducted in a DNA Thermal Cycler (Gene Amp PCR system 9600, Perkin Elmer). After an initial denaturing step at 96℃ for 1 minute, 35 cycles of PCRs were performed (94℃ for 1 minute/55℃ for 1 minute/72℃ for 1 minute), followed by a final 4 minute extension at 72℃. The PCR products were analyzed by electrophoresis in 1.5% agarose gel followed by staining with ethidium bromide. The sense and antisense primers for cIAP1 and cIAP2, respectively, were as follows: 5'-ATGCAGACACATGCAGCTCG-3' and 5'-ATGTTGGC CGCAGCATTTCC-3'; and 5'-AGGAGTCTT GCTCGTGCT GG-3' and 5'-TCCTGGGCTGTCTGATG TGG-3'. β-actin primers were used for the internal control. The sense and antisense primers for β-actin were as follows: 5'-GTG GGG CGCCCCAGGCACCA-3' and 5'-CTCCTTA/ATGTCA CG CACGATTTC-3'.
4) Transduction of adenoviruses
Adenovirus vectors expressing IκBα-SR (Ad- IκBα-SR) were kindly provided by Choon-Taek Lee (Seoul National University, Seoul, South Korea). Ad-LacZ was used as a control virus. A549 cells were transduced at multiplicities of infection (moi) of 20 by adenovirus vector in serum-free RPMI for 1 hr with gentle shaking and then washed with phosphate-buffered saline (PBS) and incubated with growth medium at 37℃, 5% CO2 incubator until use. Ad-null was used as a control virus.
RESULTS
1) Dexamethasone inhibits TRAIL and anti-cancer drug-induced cell death in A549 cells
TRAIL (100 ng/ml), taxol (100 nM), doxorubicine (1 uM), and gemcitabine (10 uM) were used in A549 cells to induce cell death. Dexamethasone (1 uM) was treated 12 hours prior to, simultaneous to, and after treatment with reagents. When A549 cells were treated with control medium, dexamethasone alone, TRAIL alone, and dexamethasone for 12 hours before the addition of TRAIL, dexamethasone inhibits TRAIL-induced cell death in A549 cells. Cell viability was assessed by light microscopy 24 hours later (Fig. 1). As well, when dexamethasone was treated simultaneously with TRAIL, TRAIL-induced cell death was inhibited. In contrast, the inhibitory cell death effect of dexamethasone was not noted when treated after TRAIL (Table 1). To evaluate the dexamethasone effect in anti-cancer drug-induced cell death, A549 cells were pretreated with dexamethasone for 12 hours before the addition of anti-cancer drugs, simultaneously treated with dexamethasone and anti-cancer drugs, and post-treated with dexamethasone for 12 hours after the addition of anti-cancer drugs. As shown in Fig. 2, the partial inhibitory cell death effect of dexamethasone was noted when dexamethasone was pretreated and simultaneously treated. It was already proven that TRAIL- and anticancer drugs induced apoptotis in various tumor cell lines. Cell death was confirmed as apoptotic by morphologic surveys with a fluorescent microscope after double staing Hoechst 33342 and propium iodide (data not shown).

Dexamethasone inhibits TRAIL-induced cell death in A549 cells. A549 cells were treated with control medium, dexamethasone alone, TRAIL alone, and dexamethasone for 12 hr before the addition of TRAIL. Cell viability was assessed by light microscopy 24 hr later.

Effect of dexamethasone and TRAIL on cell viability in A549 cells. A549 cells were treated with control medium, dexamethasone and/or TRAIL for 24 hr followed by the analysis of cell viability by crystal violet assay. In experiments with dexamethasone plus TRAIL, A549 cells were pretreated with dexamethasone for 12 hr before the addition of TRAIL, simultaneously treated with dexamethasone and TRAIL, and posttreated with dexamethasone for 12 hr after the addition of TRAIL
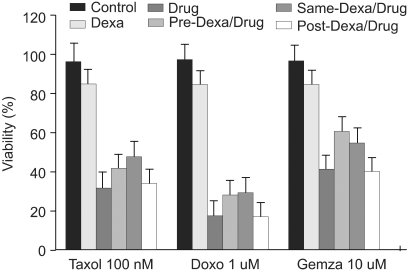
Dexamethasone inhibits anti-cancer drug-induced cell death in A549 cells. A549 cells were treated with control medium, dexamethasone and/or anti-cancer drugs: taxol, doxorubicine, and gemcitabine for 24 hr followed by analysis of cell viability by crystal violet assay. In experiments with dexamethasone plus anti-cancer drugs, A549 cells were pretreated with dexamethasone for 12 hr before the addition of anti-cancer drugs, simultaneously treated with dexamethasone and anti-cancer drugs, and posttreated with dexamethasone for 12 hr after the addition of anti-cancer drugs.
2) Induction of cIAP2 mRNA expression by dexamethasone in A549 cells
To evaluate whether dexamethasone induces cIAP mRNA expression, a member of IAP family tin regards to antiapoptotic effect, RT-PCR was performed in A549 cells. A549 cells were treated with dexamethasone and harvested at 0, 1 hr, 2 hr, and 4 hr for RT-PCR analysis with a cIAP1 primer, a cIAP2 primer, and a beta-actin primer. The result, as related to time, showed a definite increase in the level of cIAP2 mRNA. However cIAP1 mRNA expression was not affected by dexamethasone (Fig. 3).
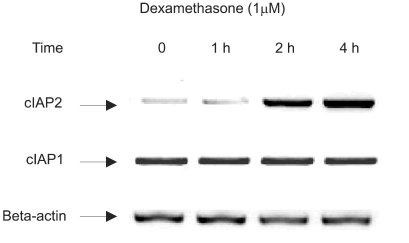
Induction of cIAP2 mRNA expression by dexamethasone in A549 cells. A549 cells were treated with dexamethasone and harvested at 0, 1 hr, 2 hr, 4 hr for RT-PCR analysis with a cIAP1 primer, a cIAP2 primer, and a beta-actin primer.
3) GR-mediated inhibitory cell death effect of dexamethasone in A549 cells
To determine whether dexamethasone inhibits TRAIL- and anti-cancer drug-induced cell death in A549 cells by GR-mediation, RU486, an inhibitor of GR, was used. A549 cells were treated with TRAIL alone, pretreated with dexamethasone 12 hours before the addition of TRAIL and pretreated with RU486 30 minutes before the addition of dexamethasone and TRAIL. As a result, the effect of dexamethasone-inhibitory cell death in A549 cells was completely reversed by RU486 (Fig. 4). This finding suggests that the anti-apoptotic effect of dexamethasone is mediated by GR activation and GR-mediated induction of anti-apoptotic genes in A549 cells. We then examined whether the induction of the cIAP2 mRNA expression is mediated by GR. A549 cells were treated with dexamethasone, pretreated with RU486 for 30 minutes prior to the addition of dexamethasone, and harvested at 0, 1 hour, 2 hours, and 4 hours for RT-PCR analysis with a cIAP2 primer and beta-actin primer. Dexamethasone-induced cIAP2 mRNA expression was not detected by RU486 pretreatment (Fig. 5).
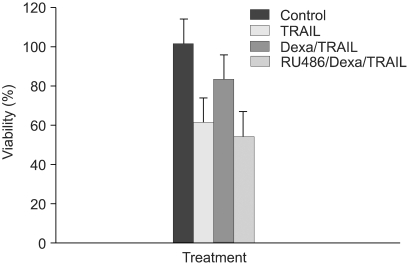
RU486 blocks inhibition of TRAIL-induced cell death by dexamethasone in A549 cells. A549 cells were treated with control medium, TRAIL, pretreated with dexamethasone 12 hr before the addition of TRAIL and pretreated with RU486 30 min before the addition of dexamethasone and TRAIL. Cell viability was assessed by crystal violet assay 24 hr later.
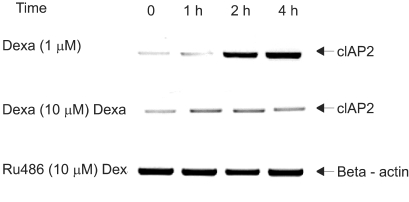
RU486 inhibits dexamethasone-induced cIAP2 mRNA expression in A549 cells. A549 cells were treated with dexamethasone, RU486 plus dexamethasone, and harvested at 0, 1 hr, 2 hr, 4 hr for RT-PCR analysis with a cIAP2 primer and beta-actin primer. In experiments with RU486 plus dexamethasone, A549 cells were pretreated with RU486 for 30 min before the addition of dexamethasone.
4) The role of NF-kB in dexamethasone-induced cIAP2 mRNA expression
We investigated whether NF-κB is involved in dexamethasone-induced cIAP2 expression because NF-κB is known to play a key role in the transcription of cIAP2. To determine whether dexamethasone-induced cIAP2 mRNA expression works through NF-κB activation, we examined the effect of NF-κB after specific inhibition of NF-κB. In order to block the activation of NF-κB effectively, IκBα mutants in which Ser32 and Ser36 were substituted with nonphosphorylatable alanine (IκBα-SR) were transduced in A549 cells. A549 cells were infected with Ad-null or Ad- IκBα-SR at 20 m.o.i for 20 hours and then incubated with TRAIL or by pretreatment for 12 hours with dexamethasone. As a result, TRAIL-induced cell death was inhibited by pretreatment with dexamethasone in both A549-Ad-null cells and A549-Ad-IκBα-SR (Fig. 6). We next examined whether the induction of cIAP2 expression could occur in A549-Ad-IκBα-SR. A549-Ad-null cells and A549-Ad-IκBα-SR cells were treated with dexamethasone and harvested at 0, 1 hour, 2 hours, and 4 hours for RT-PCR analysis with cIAP2 primer and beta-actin primer. As a result, dexamethasone-induced cIAP2 mRNA expression was increased in A549-Ad-IκBα-SR cells, as well as A549-Ad-null cells (Fig. 7). This finding suggests that the induction of cIAP2 mRNA expression by dexamethasone was through GR-mediated, but not NF-κB activation.
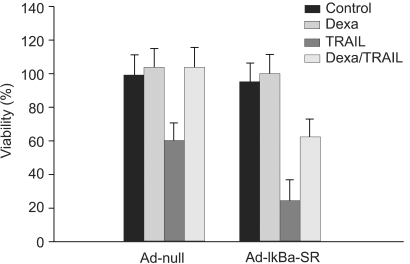
NF-κB has no effect on inhibition of TRAIL-induced cell death by dexamethasone in A549 cells. A549 cells were infected with Ad-null or Ad-IκBα-SR at 20 m.o.i for 20 hr and then incubated with TRAIL or 12 hr pretreatment with dexamethasone. Cell viability was assessed by crystal violet assay 24 hr later.
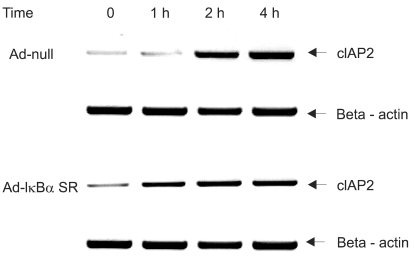
No effect of NF-κB on dexamethasone-induced cIAP2 mRNA expression in A549 cells. A549 cells were infected with Ad-null or Ad-IκBα-SR at 20 m.o.i for 20 hr, then incubated with dexamethasone and harvested at 0, 1 hr, 2 hr, 4 hr for RT-PCR analysis with cIAP2 primer and beta-actin primer.
DISCUSSION
Apoptosis is an important and complex cellular process that is highly regulated. Several signaling pathways are involved in its coordination, and vary according to cell type. Two such apoptotic signaling pathways are driven by the transcription factors NF-κB and GR. Herein we have studied the effect of GR agonists, dexamethasone and NF-κB, on one gene involved in the control of apoptosis, cIAP.
Here we have demonstrated that dexamethasone was almost a complete blocker of the TRAIL-induced apoptosis in A549 cells. In addition, dexamethasone showed a partial blocking effect on anti-cancer drugs (doxorubicine, gemcitabine, and taxol) known to induce apoptosis (16). This anti-apoptotic effect of dexamethasone was seen with pretreatment and simultaneous treatment with TRAIL or anti-cancer drugs, but was not seen when dexamethasone was given subsequent to TRAIL or anti-cancer drugs. This suggested that the anti-apoptotic effect of dexamethasone was due to a certain gene expression involved in the control of apoptosis. It was subsequently found that dexamethasone induces the transcription of cIAP2 mRNA, a member of IAP family, but did not induce cIAP1 mRNA transcription. The inhibitory effect of dexamethasone on cell death was completely reversed by pretreatment with RU486, a GR inhibitor, for 30 minutes. This suggests that the inhibitory effect of dexamethasone was mediated by GR-mediated induction of the anti-apoptotic gene, cIAP2. To confirm cIAP2 expression induced by GR and its relation to NF-κB, significant expression of cIAP2 and prevention of TRAIL-induced cell death by dexamethasone were still seen in our study when Ad-IκBα-SR was transduced in A549 cells for specific suppression of NF-κB. These results suggest that dexamethasone inhibits TRAIL- and anti-cancer drug-induced cell death in A549 cells by inducing cIAP2 gene expression through a GR-mediated, but NF-κB independent pathway.
TNF plays an important role as a cytokine in the immune system, binding to TNF receptors and causing apoptosis related to the process of cell division and growth. As the TNF family, proteins, and genetic structure were determined, it was discovered that TRAIL was a member of the TNF family, which causes apoptosis (17). Among the TNF family, TRAIL is the most similar to Fas, but is still considerably different from Fas. It causes apoptosis in cancer cells, but not in normal cells. In cancer cells, TRAIL and their R1 and R2 receptors directly activate caspases and induce apoptosis. Otherwise, normal cells have decoy receptors, R3 and R4, which work to protect normal cells from apoptosis. In several cell lines, TRAIL activates NF-κB as an anti-apoptotic transcription factor through TRAIL-R1, TRAIL-R2, and TRAIL-R4 (18). NF-κB is made of p50, p65 and c-Rel subunits and is believed to be one of the most important factors in immune and inflammatory reaction. To carry out this function, a very particular activation pathway is followed. When no stimulation existed, NF-κB binded to IκBα and IκBβ inactivation proteins, preventing movement into the nucleus. With the proper signals, however, IκBα and IκBβ become phosphorated and split NF-κB. NF-κB activation then occurs (19). TNF-α-induced apoptosis was induced by the activation of caspase via an adaptor like FADD. NF-κB activation, on the other hand, is independent of FADD and uses a different adaptor, TRAF/NIK. If NF-κB activation is inhibited in cell lines that show a resistance to TNF-α induced apoptosis, a cytotoxic effect on TNF-α carries over. A NF-κB dependent protein, cIAP2, is involved in this process. In baculoviruses, we discovered IAPs that include cIAP2 which allowed the cell to survive (20). In the human body, similar to viruses, induction of apoptosis by a variety of stimuli is suppressed by IAPs. In the IAP family, XIAP, cIAP1 and cIAP2 suppress the proteolytic activation of caspase 3, 7. Although it suppresses the activation of pro-caspase 9, it does not suppress the activation of caspase 1, 6, 8 or 10. NF-κB is stimulated by TNF-α, induces the expression of cIAP2. This induced cIAP2 suppresses the death of the cell by inactivating caspase.
It can be observed that glucocorticoids cause an anti- inflammatory and immunosuppressant effect by strongly inducing apoptosis in immunologic cells, such as T-cells and monocytes. Activated cytotoxic T-cells excrete cytokines, such as INF-γ and TNF-α, and stimulate the apoptosis pathway, causing apoptosis or removing damaged epithelial lung cells. In the past, epithelial lung cells were thought to be only a physical barrier against the outside environment, but it has recently been determined that they are involved in the inflammatory process by excreting inflammatory mediators, such as TNF-a, IL-1B, IL-6, chemokines such as IL-8, RANTES and prostaglandin E2, endothelin, and similar inflammatory mediators., Epithelial lung cells have been widely recognized as secretors of GM-CSF, which increase the inflammatory effect. In A549 epithelial lung cells, dexamethasone induces cIAP2 expression and suppresses INF-γ-, anti-FAS-induced apoptosis. This is brought about through suppression of caspase 3 and 7 activation (21). In another study, TNF-α increased glucocorticoid-induced transcription of GR. It also increased the amounts of GR and GRE binding GR (22). The results of this study show that IFN-γ enhances GR activation in A549 epithelial lung cells. This causes dexamethasone to increase the expression of cIAP2. TNF increases the expression of cIAP2 mRNA. Within the cIAP2 promoter, there are two NF-κB binding sites controlled by TNF (14). In another study, dexamethasone increases GR-dependent cIAP2 promoter transcription via GRE, but at NF-κB binding sites it does not suppress TNF-induced promoter activation (15). In epithelial lung cells, glioma cells, and T-cells, cIAP2 expression by dexamethasone is related to the suppression of apoptosis induced by anti-FAS antibody. It has been reported that in MCF-7 carcinoma cells, dexamethasone suppresses cIAP1, cIAP2, and XIAP expression, which causes the suppression of TNF-α and induces apoptosis.
Presently, NF-κB is perceived to be the most important transcription factor of cIAP expression. In fact, the cIAP2 gene promoter region has two NF-κB binding regions (23). However, dexamethasone is the most significant NF-κB inhibitor (24). That is, cIAP2 expression is induced by NF-κB, but on the other hand, the NF-κB inhibitor, dexamethasone induces the expression of cIAP2. This appears to be a contradiction. Thus, in order to understand tthe problem of the mechanism of GR-mediated cIAP2 expression, an understanding of the role of NF-κB is crucial. In order to understand the relation of dexamethasone-induced cIAP2 expression and NF-κB in our study, we transduced Ad- IkBa-SR in A549 epithelial lung cells for the specific inhibition of NF-κB. As a result, we found that dexamethasone still induced cIAP2 expression and inhibited cell toxicity. That is, dexamethasone, a synthetic glucocorticoid, inhibits TRAIL- and anti-cancer drug-induced cell death through cIAP2 expression by a GR-activated, but NF-kB independent pathway in A549 lung epithelial cells. TNF-α, IFN-y, IL-1β, IL-6, IL-8, GM-CSF, PMA, and a reactive oxygen metabolites are stimulators of NF-κB activation. Most of these genes are activated by NF-κB, which is related to immunologic and anti-inflammatory effects. These genes are mostly suppressed by glucocorticoids, so the suppression of NF-κB activation is the most important mechanism in regards to anti-inflammatory effect. Initially, it was known that the mechanism of NF-κB inhibition of glucocorticoids was the induction of IκBα transcription (24). However, this occurs only in T-cells, monocytes, and HeLa cells. Subsequent studies of vascular endothelial cells showed that the NF-κB suppression of glucocorticoids and IκBα were not related. In reality, apart from immunologic cells, inhibition of NF-κB activation through protein-protein interaction exists between GR and NF-κb in most cells (23).
The results of our study confirmed that the NF-κB, which was believed to have an important role in cIAP2 expression, is not involved in dexamethasone and inhibits TRAIL- and anticancer drug-induced cell death in A549 epithelial lung cells. Therefore, how is glucocorticoids related to cIAP2 expression? Using this information, it was confirmed in our study that inducing cIAP2 expression through GR-mediated by dexamethasone in A549 lung epithelial cells is a new provision of the control mechanism of cIAP2 expression confined to NF-κ B. In fact, the cIAP2 gene promoter was shown to have a GRE, as a GR binding region (14). Also, dexamethasone stimulates transcription from the cIAP2 promoter region in a GR-dependent manner through a GRE, and did not repress TNF-induced promoter activity at the NF-κB binding region (15). In the future, further study is need regarding activation of cIAP2 promoters by dexamethasone. Apoptosis inhibition by induction of Bcl-2 and Bcl-XL expression has also been reported. Other methods of induction of cIAP2 expression can explain the further new mechanisms of the anti-apoptotic effect of dexamethasone.
Glucocorticoids are an important drug for the treatment of ARDS and pulmonary fibrosis. These diseases have been recently observed to be related to apoptosis of epithelial lung cells. Therefore, much attention has been paid to the pathophysiology of these diseases (25). Dexamethasone has contradicting effects in regards apoptosis. It caused apoptosis in immunologic cells, but it suppresses apoptosis in epithelial lung cells. Biologically, induction of apoptosis in activated immunologic cells reduces inflammation and inhibition of apoptosis in epithelial lung cells, decreasing cell toxicity and reducing lung damage. It is believed that dexamethasone will give a protective effect with radiotherapy and anti-cancer drug therapy for epithelial lung cells, but further study is needed using primary lung cells, as well as other means.
CONCLUSIONS
Dexamethasone inhibits TRAIL- and anti-cancer drug-induced apoptosis in A549 cells by inducing cIAP2 gene expression through a GR-mediated, but NF-κB-independent pathway.
Notes
This work was supported by a Korea Research Foundation Grant. (KRF-2001-F00131)