A Histone Deacetylase Inhibitor, Trichostatin A, Enhances Radiosensitivity by Abrogating G2/M Arrest in Human Carcinoma Cells
Article information
Abstract
Purpose
Histone deacetylase inhibitors (HDIs) are emerging as potentially useful components in anticancer therapy. In this study, we tried to confirm the radiosensitizing effect of trichostatin A (TSA) on a panel of human carcinoma cell lines and elucidate its mechanism of interaction.
Materials and Methods
A549, HeLa and Caski cells were exposed to TSA for 18 hr prior to irradiation, and the cell survival then measured using a clonogenic assay. Western blot and flow cytometric analyses, for histone acetylation, and cell cycle and apoptosis, respectively, were also performed.
Results
TSA increased the acetylation of histone H3. The pretreatment of TSA consistently radiosensitized all three cell lines. The SF2 (surviving fraction at 2 Gy) of TSA-treated cells was significantly lower than that of mock treated cells. The SER (sensitizer enhancement ratio) increased in all 3 cell lines, in concentration dependent manners. The TSA treated cells showed abrogation of radiation-induced G2/M arrest, in a concentration dependent manner.
Conclusion
The pretreatment of TSA enhanced the radiosensitivity of a panel of human carcinoma cells, which was attributed, in part, to the abrogation of radiation-induced G2/M arrest.
INTRODUCTION
DNA is packaged into nucleosomes, the repeating units of chromatin, which is composed of -146 base pairs of two superhelical structure turns of DNA wrapped around an octamer core of histone pairs; H2A, H2B, H3 and H4. The post-translational modifications in gene regulation include: acetylation of lysines, methylation of lysine and phosphorylation of serine. Accumulation of acetylated histones is associated with neutralization of the lysine-positive charge, which leads to an alteration in chromatin conformation, and may provide for greater access to the promoter regions of genes for transcription factor complexes. The pattern of acetylation of histone, as well as those of methylation and phosphorylation, may represent a "code" that is recognized by the nonhistone complexes involved in the regulation of gene expression (1~3). HDACs also target nonhistone proteins, such as transcriptional factors (p53), nuclear receptors (gluticorticoid and estrogen receptor) and cell cycle regulation proteins (pRb). The acetylation of these non-histone cellular proteins is thought to modulate their activity (4~6).
Histone deacetylase inhibitors (HDIs) are involved in various biological effects, including transcriptional regulation, cell differentiation, cell cycle arrest, anti-angiogenesis and apoptosis (2,7,8). The HDAC-Is are known to have in vitro and in vivo anti-tumor activity against transformed cells of various histological origins (1,3), and some HDIs already have been tested in clinical trials (9~11).
The anti-tumor effect of HDIs has been extensively investigated, while in contrast, only a few studies have reported the radiosensitizing effect of HDIs in human malignant cell lines (12~17). Those studies have demonstrated several structurally unrelated HDIs to have an in vitro radiosensitizing effect. With regard to the potential application of HDIs in the treatment of solid tumors, an important question is whether the treatment efficacy would be influenced by the intrinsic differences between cancer cells, such as the different histologies and sites of origin. Another important question pertains to the elucidation of the mechanisms underlying HDI-induced radiosensitization, which remain to be studied.
In this study, we attempted to elaborate on our previous studies (17) by testing the radiosensitizing effect of TSA in other carcinoma cell lines of different histological origins, and understand the mechanism of interaction. We found that pretreatment with TSA consistently radiosensitized a panel of tested human carcinoma cell lines, which was attributed, in part, to the abrogation of radiation induced G2/M arrest.
MATERIALS AND METHODS
1) Cell culture
The A549, HeLa and Caski cell lines were obtained from the Korean Cell Line Bank. Cells were cultured at 37℃ in water saturated with 5% CO2. Cultures were maintained in DMEM (Welgene, Daegu, Korea) or RPMI media (Gibco, Grand Island, NY), supplemented with 10% fetal bovine serum and 12.5 µg/ml gentamicin (Gibco), respectively.
2) Pharmacologic inhibitor
The TSA was obtained from Sigma Chemical Co. (St. Louis, MO), and dissolved as concentrated stock solutions in DMSO, stored at -20℃ and diluted in the respective culture media at the time of use. Control cells were treated with the media containing an equal concentration of the drug carrier, DMSO.
3) Clonogenic assays
A specified number of cells were seeded into each well of six well culture plates, and treated with 50, 100, 200 and 400 nM TSA as the HDI. After exposure for 18 hours, the cells were irradiated with 4 MV X-ray from a linear accelerator (Clinac 4/100, Varian Medical Systems, Palo Alto, CA), at a dose rate of 2.46 Gy/min, and then incubated for 14 to 21 days to allow colony formation. The colonies formed were fixed with methanol, stained with 0.5% crystal violet, the number of colonies containing at least 50 cells determined, and the surviving fraction then calculated. The survival data were fitted to a linear-quadratic model using a nonlinear regression within the JMP5.0.1a software (SAS Institute Inc. Cary, NC). Each point on the survival curves represents the mean surviving fraction from at least three dishes. Comparisons between the SF2 of the TSA- and mock-treated cells were performed using the t test within the SAS software. The sensitizer enhancement ratio (SER) was defined as the ratio of the isoeffective dose at SF 0.5, in the absence compared to the presence of TSA. The SF values are presented as the mean of values of triplicate experiments.
4) Western analysis
Cells were washed and scraped, and then resuspended in lysis buffer (iNtRON Biotechnology, Seoul, Korea). The proteins were solubilized by sonication, and equal amounts separated on SDS-PAGE and then electroblotted onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA). The membranes were blocked with PBS containing 0.1% Tween 20 and 5% powdered milk, and then probed with the primary antibody directed against polyclonal rabbit anti-acetyl-histone H3 IgG (Upstate, Lake Placid, NY), at a 1:1,000 dilution, and the monoclonal anti-α-tubulin antibody (Sigma, St. Louis, MO) at a 1:5,000 dilution. The membranes were washed, and then incubated with secondary antibodies, consisting of peroxidase-conjugated goat anti-rabbit or mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, at a 1:2,000 dilution, for 1 hour. Detection of antibody binding was performed using ECL detection kits from Amersham, with the appropriate secondary antibody supplied with the kit.
5) Flow cytometric analysis
A549 cells were treated with 50, 100, 200, 600 or 1,000 nM TSA for 6~72 hr. The cells were then trypsinized, washed and fixed at a concentration of 2~3×106 cells/ml in a PBS suspension, containing 2.0 ml 1% formaldehyde, for 15 min. After centrifugation at 12,000 rpm for 5 minutes, the cell pellets were resuspended in ice-cold 80% ethanol for 24 hr for fixation. The fixed cells were then washed twice with PBS, suspended in 1 ml 0.25% Triton X-100 (Merck) in PBS on ice for 5 min, centrifuged, washed, and resuspended in PBS containing 5 µg/ml propidium iodide (Molecular Probes, Eugene, OR) and 0.1% RNase A (Sigma). To analysis for any cell cycle change after the irradiation, A549 cells were pretreated with 0, 200 or 400 nM TSA for 18 hr, and then irradiated with 8 Gy. At 2~24 hr after irradiation, the cells were collected, fixed and stained with propidium iodide, and then analyzed. At least 1×104 events were counted.
RESULTS
1) TSA increased acetylation of histone H3 and increased radiosensitivity
The TSA increased the acetylation of histone H3, a presumptive useful marker of the biological activity of HDIs (Fig. 1). Pretreatment with TSA consistently radiosensitized all three cell lines, although the level of radiosensitization varied depending on the individual cell line tested (Fig. 2A, B and C). The SF2s of the TSA-treated cells were significantly lower than those of the mock treated cells in all three cell lines. The SF2s of A549 cells decreased significantly after pretreatment with 100 nMTSA, and those of the HeLa and Caski cells were significantly decreased by treatment with 200 nM TSA, as shown in Fig. 3 (p<0.05). The SERs of all three cell lines increased in concentration dependent manners; the increase in the SER of the A549 cells reached at plateau at 100 nM TSA (Fig. 4).
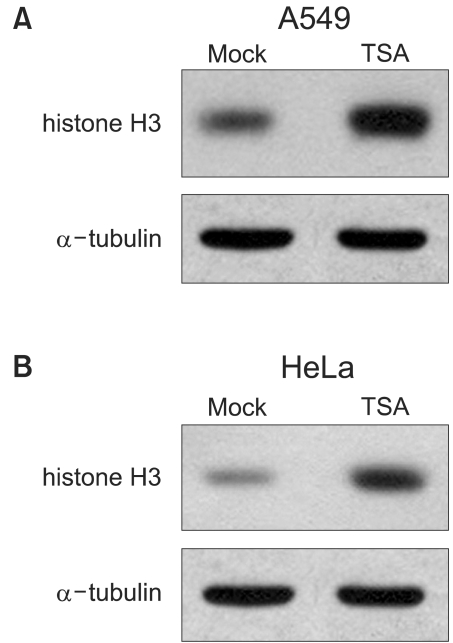
TSA increased hyperacetylation of histone H3. Cells were treated 200 nM TSA for 18 hr, the proteins harvested, separated on SDS-PAGE and electroblotted. The membrane was probed with anti-histone H3 antibody. α-tubulin was used as a loading control. The same amount of drug carrier (DMSO) treated cells were used as a mock control. (A) A549; (B) HeLa.

Survival curves for cells treated with trichostatin A (TSA) prior to irradiation. Cells were pretreated with 0, 50 or 200 nM TSA for 18 hr, and then irradiated with 4 MV X-rays after the removal of the TSA from the culture media. The survival was measured using a clonogenic assay. Each point on the survival curves represents the mean surviving fraction from at least three dishes. Experiments were repeated a minimum of three times. The error bars represent the standard errors. (A) A549; (B) HeLa; (C) Caski.

Surviving fraction of cells pretreated with trichostatin A (TSA) prior to 2 Gy (SF2) of irradiation. Cells were pretreated with 0, 50, 100 or 200 nM TSA for 18 hr, and then irradiated with 4 MV X-rays after the removal of the TSA from the culture media. The survival was measured using a clonogenic assay. The SF2s of the TSA-treated cells were compared with those of mock treated cells using the t test. The error bars represent the standard errors.
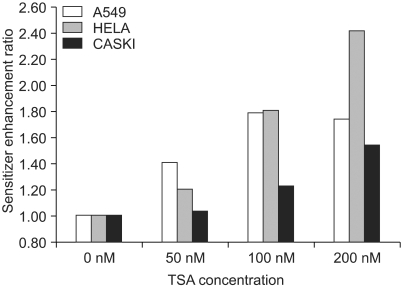
Sensitizer enhancement ratio (SER) of trichostatin A (TSA). The SER was defined as the ratio of the radiation dose required to obtain a surviving fraction (SF) of 0.5, without TSA pretreatment, to that required to obtain the same SF after TSA pretreatment. Cells were pretreated with 0, 50, 100 or 200 nM TSA for 18 hr, and then irradiated with 4 MV X-rays after the removal of the TSA from the culture media. The survival was measured using a clonogenic assay.
2) Effect of TSA on cell cycle distribution and apoptosis
In the A549 cells, the proportion in the G2/M phase was steeply increased when the cells were exposed to concentrations higher than 200 nM, and reached a plateau (Fig. 5). The subG1 proportion, as a presumptive indicator of apoptosis, started to increase when the cells were treated with concentrations higher than 200 nM TSA for more than 12 hours. However, a significant amount of apoptosis was detected only when the cells were exposed to concentrations higher than 600 nM for longer than 24 hrs (Fig. 6).
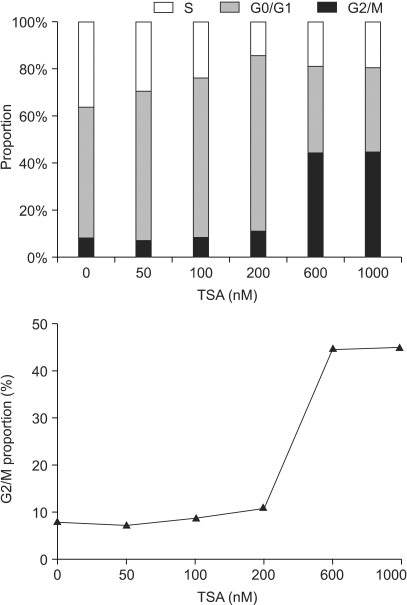
Cell cycle analysis for A549 cells treated with trichostatin A (TSA). Cells were treated with 0~1,000 nM TSA for 18 hr, and collected, fixed and stained with propidium iodide, and then analyzed using flow cytometry.

Sub-G0/G1 proportion in A549 cells pretreated with trichostatin A (TSA). Cells were treated with 0~1,000 nM TSA for 6~48 hr, and the cell cycle distribution then determined using flow cytometry. The proportions of cells with DNA contents less than that in the G0/G1 phase cells were defined as the sub-G0/G1 proportions.
3) Cell cycle progression following irradiation in the cells pretreated with TSA
The mock-treated control cells were accumulated in the G2/M phase after irradiation, and in a radiation dose dependent manner, in terms of the magnitude and duration (Fig. 7A). A brief episode of moderate G2/M arrest was observed, with a peak at 6 hours, which returned to the basal level at 10 to 12 hours following irradiation at the dose of 2 Gy, but the G2/M proportion was increased by less than 20% of the basal level (Fig. 7B, -○-). For cells irradiated at doses higher than 8 Gy, a longer episode of robust G2M arrest was noted; the G2/M proportion rapidly increased to more than 40% of the basal level, showed a peak at 12 hours, but did not return to the basal level until 24 hour after the irradiation (Fig. 7C, -○-).
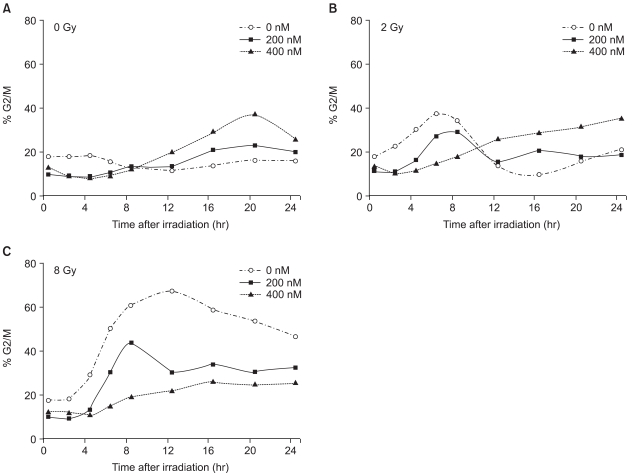
The change in the G2/M proportions of cells following the combination of TSA and irradiation. A549 cells were pretreated with 0 nM (-○-), 200 nM (-□-) and 400 nM(-△-) TSA for 18 hr. The TSA was then removed from the media, and the cells irradiated with 4 MV X-rays at 0 Gy (A), 2 Gy (B) or 8 Gy (C) at 0 hr. After irradiation, the cells were collected, fixed and stained with propidium iodide, and then analyzed using flow cytometry at 2, 4, 6, 8, 12, 16, 20 and 24 hr.
In contrast to the mock treated cells, the TSA treated cells showed abrogation of the radiation-induced G2/M arrest, in a concentration dependent manner. When the cells were treated with 200 nM TSA, the magnitude of radiation-induced G2/M arrest significantly decreased (Fig. 7B and C, -□-). In the case of the 400 nM TSA treated cells, the abrogation of G2/M arrest was more evident and robust (Fig. 7B and C, -△-).
DISCUSSION
There is no doubt that in the relatively short time since the discovery of HDACs great progress has been made, both in the understanding of mediators of gene silencing, as well as in the translation of this knowledge into potential clinical use. This study showed that the pretreatment of cells with TSA enhanced the cellular radiosensitivity. Both this and our previous study (17) support the notion that radiosensitization by TSA is a rather general phenomenon. Other investigators have also reported HDI-induced radiosensitization (12~16). In view of the fact that HDIs with different chemical structures have a common radiosensitizing effect, it would appear that HDAC is involved in the common cellular response to ionizing radiation. In this study, TSA consistently radiosensitized all three cell lines we tested, although the level of radiosensitization varied in terms of the SF2s and SERs. Dose-response relationships were clearly observed by the dependence of the SERs on the TSA concentration. This observation indicates that the level of radiosensitization might depend on the intensity of the HDAC inhibition; even though TSA mediated radiosensitization is a general phenomenon. There have been other reports of a similar concentration-dependent radiosensitization using sodium phenylbutyrate (13) and TSA (14).
The proportion of cells in the G2/M phase from our data was not significantly changed by pretreatment with 200 nM TSA for 18 hr, which is with other reports where HDIs, such as TSA, arrested the cell cycle progression in the G2/M phase. It seems that the cellular radiosensitivity might be enhanced at much lower concentrations than those that induce the cell cycle arrest and G2/M accumulation, which is consistent with the results of other investigators, who reported the accumulation of cells in the G2/M phase only after exposure to 1 mM or higher concentrations of TSA (18). Admittedly, it is possible that our analysis of the effects of TSA on the cell cycle distribution might have been confounded by TSA-induced cell death, especially at concentrations above 200 nM. Apoptosis and cell cycle arrest are regarded as the major mechanisms underlying the anti-tumor activity of HDIs. Our results show that apoptosis was significantly detected only when cells were exposed to concentrations higher than 600 nM and for longer than 24 hrs. A substantial number of studies have shown that various HDIs consistently induce apoptosis. Setting aside the effects of the different methodologies used to measure apoptosis, it seems that HDI pretreatment has a more intense effect than was observed under our experimental conditions. In the case of TSA, the majority of these other studies pretreated cells with 600~1,000 nM TSA and for longer than 24 hr (19). Our results, and those of other studies, on TSA-induced G2/M arrest, suggest that TSA enhances the cellular radiosensitivity at much lower concentrations than those required for anti-tumor activity. It is not yet known whether the radiosensitization caused by HDIs and their anti-tumor activities share common intracellular pathways.
Camphausen et al reported the number of cells expressing γ2AX foci was significantly greater in MS-275 treated cells at 24 hour after irradiation. This result indicates that MS-275 induced radiosensitization may involve an inhibition of DNA repair (15). The exposure of cells to ionizing radiation has been reported to delay the normal progression through the cell cycle on several of the checkpoint that exist in the cell cycle, and these play an important role in the preservation of the genomic integrity in cells with damaged DNA (20). Of these, G2 arrest in irradiated cells is implicated as a determining factor of cellular radiosensitivity (21). Once considered as a passive consequence of DNA damage, G2 arrest is actually an active response, with a role in DNA repair. ATM and ATR are thought to direct DNA damage signals down to the G2 checkpoint machinery level, which arrests irradiated cells. Thus, by inhibiting ATM- or ATR-associated HDACs, HDIs might determine the radiation-induced activation of the G2 checkpoint (22). HDAC4, one of the class II HDACs, is a critical component of the DNA damage response pathway that acts through 53 BP1, one of two proteins that interact with the transactivation domain of p53, which abrogates the DNA damage-induced G2 delay and radiosensitized the HeLa cells (23). This reflects the crucial role of HDACs in radiation-induced G2 checkpoints. In this study, the HDAC inhibition was observed to abrogate the accumulation of irradiated cells in the G2/M phase, which is an important mechanistic clue that underlies TSA-induced radiosensitization.
The aforementioned findings suggest that HDACs are extensively involved in the cellular response to ionizing radiation, which means they provide potential molecular steps that can be targeted by HDIs at multiple levels of the cellular response to radiation-induced DNA damage. It is known that HDACs, in the context of their role in the DNA damage response, may be targeted at some specific steps between DNA damage recognition and the G2/M checkpoint. However, precise detailed mechanisms of the radiosensitization caused by HDIs remain to be defined. Our mechanistic study focused on the mitotic check point machinery, and is ongoing. Based on the implication that HDAC might be a potential target for the modulation of non-histone molecules, we are also looking at the relevant change in non-histone proteins related to radiosensitivity, which may correlate with our observation.
In view of the potential clinical application of HDIs, one key question relates to which of the HDACs are important for the inhibition of the desired pharmacological profile to obtain a useful anticancer therapy. One important characteristic of HDIs is their selectivity in altering the gene expression in transformed cells. The available compounds induce accumulation of hyperacetylated histones in most chromatin regions, but only a small subset of expressed genes show a change in their transcription patterns (24). The treatment of normal and tumor cells with HDIs causes similar accumulation of acetylated histones. Nevertheless, tumor cells appear to be much more sensitive than normal cells to the growth inhibition and apoptotic effects of HDIs (25). This could be a very meaningful observation for a potential strategy for the use of HDIs in a clinical setting. As with any other anticancer agent, the drugs that target the chromatin structure also face the problems of their specificity of action. Given the large number of combinatorial possibilities with this specific gene regulation exist and could be exploited as targets with the use of drugs developed to match such specificities.
CONCLUSIONS
HDI enhances the radiation induce cell killing in the various cancer cells having intrinsic difference, which may serve as a general strategy for enhancing the radiosensitivity of tumor cells. The abrogation of radiation-induced G2 arrest seems to be an important mechanistic clue for TSA-induced radiosensitization; however, further detailed studies are ongoing. These results have potential implications for the clinical utility of HDIs in increasing the anticancer efficacy of radiation.
Notes
Supported by grant Nos. 04-2004-47 & 03-2005-009 from the Seoul National University Hospital Research Fund (IHK) and by grant No. 800-20040311 from the Seoul National University Fund (JSK).