Utilizing Plasma Circulating Tumor DNA Sequencing for Precision Medicine in the Management of Solid Cancers
Article information

Abstract
Plasma circulating tumor DNA (ctDNA) sequencing has demonstrated clinical utility for tumor molecular profiling at initial diagnosis or tumor progression in advanced solid cancers and is being rapidly incorporated into the clinical practice guidelines, including non–small cell lung and breast cancer. Despite relatively low sensitivity, plasma ctDNA sequencing has several advantages over tissue-based assays, including ease of sampling, rapid turnaround time, repeatability, and the ability to overcome spatial heterogeneity, which makes it ideal for investigating acquired resistance and monitoring tumor evolution and dynamics. With technological advancement and declining costs, the clinical application of plasma ctDNA is expanding, and numerous ongoing clinical trials are examining its potential to guide the management of advanced, localized, and even preclinical cancers of various tumor types. The ability of plasma ctDNA analysis to detect minimal residual disease following curative treatment in the absence of clinical disease is among its most promising attributes. Plasma ctDNA sequencing can also facilitate the conduct of clinical trials and drug development, particularly in immunotherapy. In order to incorporate plasma ctDNA sequencing for clinical decision-making, it is important to understand the preanalytical and analytical factors that may affect its sensitivity and reliability.
Introduction
Identifying tumor molecular profiles, particularly multigene panel assays based on next-generation sequencing (NGS), has become essential for the management of many solid tumors [1–3]. While tissue-based assays have been the gold standard for mutational profiling of tumors, circulating tumor DNA (ctDNA) testing has emerged rapidly as a new diagnostic tool that can supplement or replace tissue-based assays [4]. CtDNA analysis is a subset of “liquid biopsies,” a broader category that includes the analysis of circulating extracellular nonencapsulated nucleic acids (DNA, RNA, and miRNA), tumor cells, proteins, nucleosomes, and extra-cellular vesicles, in body fluids, such as blood, urine, stool, ascites, pleural fluid, saliva, or cerebrospinal fluid [5]. Although various liquid biopsy techniques for tumor molecular profiles have been studied to date, plasma ctDNA assays have been evaluated most extensively to be considered in the standard of care (SoC) [4,6–8].
Circulating cell-free DNA (cfDNA) refers to free-floating, fragmented molecules derived from various cells in the human body (primarily through apoptosis or necrosis) and released into circulation and detected in the noncellular (“cell-free”) component (e.g., plasma) of the blood or other body fluids [5,9]. In healthy individuals, plasma cfDNA concentration ranges from 0 to 100 ng/mL and is primarily derived from hematopoietic cells. However, in cancer patients, additional DNA shed by tumor cells (ctDNA) is released into the bloodstream and added to the cfDNA already present. Therefore, the concentration of cfDNA in cancer patients can range from 0 to greater than 1,000 ng/mL [10,11]. The fraction of ctDNA in total plasma cfDNA depends on multiple factors, including tumor types, histology, stages, locations, vascularity, and concurrent interventions (surgery or chemotherapy), and is highly variable between patients, ranging from less than 0.01% to greater than 90% [12]. Plasma cfDNA fragment size is primarily distributed around 167 or 320 bp, but can range > 10,000 bp, indicating the involvement of multiple mechanisms in the release of cfDNA into the blood [10,12]. Compared to cfDNA, ctDNA is characterized by shorter fragment size (90–150 bp), though the underlying cause of this disparity remains unclear [13,14].
Since the amount of ctDNA in the blood of cancer patients is typically very low, high sensitivity is the primary requirement for analyzing plasma ctDNA. Digital polymerase chain reaction (PCR) techniques, such as BEAMing (beads, emulsion, amplification, and magnetics) or droplet digital PCR, can detect variants of variant allele frequency as low as 0.01% in cfDNA, and have thus been extensively utilized for plasma ctDNA genotyping [15,16]. Although digital PCR-based techniques are highly sensitive, they can only analyze a small number of known variants in a single assay. In contrast, NGS-based techniques enable comprehensive molecular profiling of plasma ctDNA by interrogating from several hundreds of variants (targeted panel sequencing) to the entire exome or genome in a single experiment. NGS-based ctDNA analysis was initially less sensitive than digital PCR-based techniques due to amplification/sequencing errors and required more DNA input. However, deeper sequencing coverage coupled with a continuous decrease in NGS cost, as well as molecular barcoding methods (each single input DNA fragment is tagged with a unique molecular barcode before amplification/sequencing) and error suppression bioinformatics pipelines has significantly improved the sensitivity of NGS to be comparable to or even superior to digital PCR.
This review will focus on plasma ctDNA sequencing in the management of solid tumors. The NGS-based plasma ctDNA assay has been extensively evaluated in cancer patients and is becoming more prevalent as a result of its ongoing technical improvements. Its clinical benefit has been established and integrated into clinical practice for several indications. Aside from that, it has a substantial future clinical utility and application potential. Plasma ctDNA sequencing studies have primarily emphasized its clinical utility in permitting serial monitoring of tumors, thus elucidating tumor evolution over time (temporal heterogeneity). Plasma ctDNA sequencing can capture molecular portraits of multiple metastatic tumors in a single assay and has the potential to overcome intra-lesional or spatial heterogeneity compared to tissue sequencing. This is because ctDNA is derived from multiple metastatic sites, with each typically contributing only a small portion of total ctDNA. The majority of plasma ctDNA sequencing studies employed a targeted panel approach and did not maximize its potential. A more thorough evaluation of ctDNA, including whole-genome sequ-encing (WGS), could reveal its actual role in cancer. This is illustrated by a study using deep WGS of ctDNA in patients with treatment-resistant metastatic prostate cancer [17].
In this review, we will first discuss the clinical application of plasma ctDNA sequencing in advanced solid cancers and then move on to localized or preclinical cancers (Fig. 1). Practically, the clinical application of plasma ctDNA in patients with solid cancers can be divided into a qualitative assessment of tumors to identify molecular alterations to guide treatment in a given cancer and a quantitative assessment of tumors to diagnose microscopic cancer prior to clinical diagnosis or to monitor tumor burden throughout the patient’s cancer journey. Although this classification may be ambiguous and may overlap in many instances, we will attempt to cover the qualitative and quantitative aspects of plasma ctDNA sequencing separately in each section. Additionally, the role of plasma ctDNA in the clinical trial and practical considerations when sequencing plasma ctDNA in patients with solid cancers will be discussed.
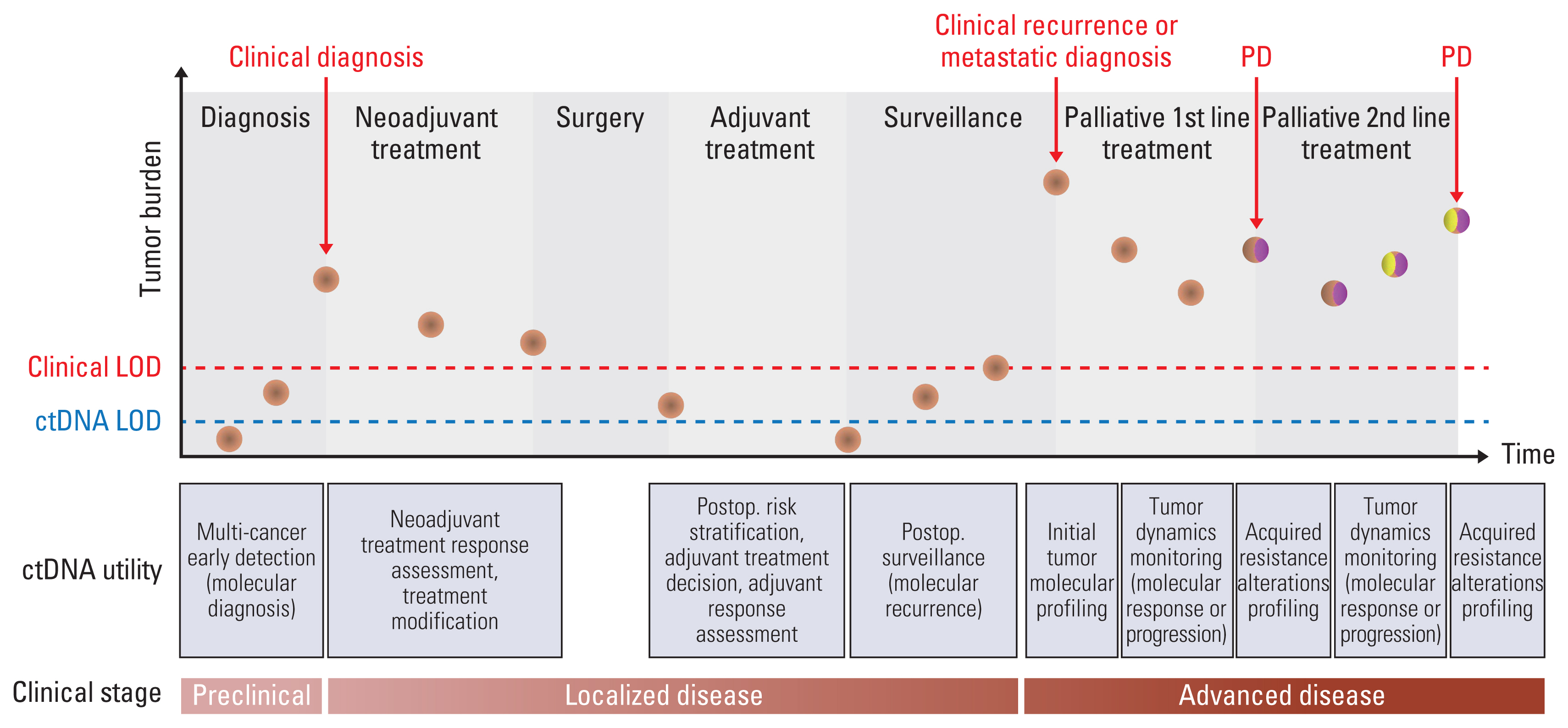
The clinical utility of plasma circulating tumor DNA (ctDNA) sequencing for the management of solid tumors in various clinical scenarios. At each time point, the brown circles represent a major clone within the tumor. Yellow and burgundy ellipses within brown circles represent the development of subclones in tumors at disease progression (PD) following first- and second-line therapy. The limit of detection (LOD) for clinical modalities and plasma ctDNA sequencing is represented by the red and blue dashed lines, respectively.
Clinical Application of Plasma ctDNA Sequencing in the Management of Advanced Solid Cancers
1. Initial molecular profiling at diagnosis of recurrent or metastatic solid cancers
NGS-based molecular profiling of tumors for therapy selection has become an integral part of the initial diagnosis of advanced or metastatic tumors for numerous solid cancers, such as non–small cell lung cancer (NSCLC), prostate, ovarian, and cholangiocarcinoma.
Although tissue-based multigene assays are still the gold standard for molecular profiling of solid cancers, plasma ctDNA sequencing eliminates the need for invasive biopsies over tissue-based assays, making it an attractive alternative for patients who are unable to undergo invasive sampling due to medical conditions or tumor location, or who have insufficient or poor-quality biopsy tissue [1–3,18]. In addition, while tissue-based assays usually have a longer turnaround time (TAT) of up to 4 weeks, the TAT for plasma ctDNA sequencing was significantly shorter than tissue-based assays (median, 7–9 vs. 15–19 days) in multiple studies. The short TAT with plasma ctDNA assay could potentially reduce delays in treatment initiation and could benefit patients who require prompt treatment decisions, such as those with aggressive cancer [19–21]. For these subsets of patients, plasma ctDNA sequencing can be recommended in the initial tumor profiling as a “parallel” (concurrent tissue and ctDNA assay), or “plasma first” approach (ctDNA assay followed by selective tissue testing).
Other than the aforementioned indications, tissue-based sequencing is currently the preferred method for initial tumor profiling. Nevertheless, given the recently demonstrated high concordance rates between ctDNA and tissue assays, plasma ctDNA sequencing may be considered for use in clinical practice. A validated comprehensive ctDNA sequencing (Guardant360) demonstrated high sensitivity (80%) and concordance rate (> 98.2%) for guideline-recommended biomarkers in NSCLC [21], as well as a high concordance rate (89.9%–100%) in advanced gastrointestinal cancer [19,22]. Also, 96%–99% and 90%–93% concordance rates with ctDNA digital PCR and tissue sequencing were reported for breast and colorectal cancer (CRC), respectively [20,23–25]. Although tissue genotyping is the SoC, plasma ctDNA can be considered with caution as the initial method for tumor profiling, according to these findings. This is particularly pertinent for oncogene-dependent NSCLC, and a recent consensus statement from the International Association for the Study of Lung Cancer recommended that liquid biopsy is an acceptable option as the initial approach (“plasma first”) for patients with oncogene-driven NSCLC at the time of diagnosis [8].
Importantly, plasma ctDNA sequencing may be complementary to tissue-based assays for initial tumor profiling in at least some clinical scenarios, even when sufficient tissue is available for sequencing. Although tissue sequencing confirmed wild-type RAS in metastatic CRC patients prior to first-line anti–epidermal growth factor receptor (EGFR) therapy, the presence of RAS or PIK3CA mutations in baseline ctDNA was associated with significantly worse outcomes [26–28]. This suggests that plasma ctDNA may be able to detect more patients with resistant mutations that were missed by tissue-based testing due to tumor heterogeneity or sensitivity issue with tissue assay, thereby enhancing the selection of patients for targeted therapy.
When implementing plasma ctDNA sequencing for tumor profiling, it is important to keep in mind the relatively low sensitivity (higher false-negative rate) of plasma ctDNA sequencing compared to tissue sequencing. The sensitivity and specificity of plasma ctDNA assays are highly dependent on the concentration of ctDNA. Therefore, negative findings with plasma ctDNA sequencing should not be interpreted as reliable when the ctDNA concentration is lower than the limit of detection of the assay, and tissue confirmation may be required in such cases. Moreover, plasma ctDNA assays have a lower sensitivity for structural variations (SVs), such as fusions or copy number alterations, when compared to tissue-based assays. Although a recent study found that pertuzumab and trastuzumab had comparable efficacy in metastatic CRC patients with human epidermal growth factor receptor 2 (HER2) amplification in tissue versus ctDNA [29], further validation studies are required to recommend plasma ctDNA sequencing as the primary method for SVs detection outside of clinical trials.
In addition to somatic mutations, plasma-based microsatellite instability evaluation exhibits high overall agreement rates of 94% to 98.2% with tissue testing and can be incorporated into tumor profiling to direct treatment with an immune checkpoint inhibitor (ICI) [30,31]. High tumor mutation burden (TMB) is another biomarker recognized by the US Food and Drug Administration for ICI therapy in all solid tumors. Despite the fact that TMB is calculated using tissue, blood-based TMB (blood TMB or bTMB) has shown promise in predicting ICI benefits in retrospective studies. In the recent prospective phase 2 B-F1RST study, a cutoff score of 16 on the Foundation Medicine bTMB assay was associated with a higher overall response rate (ORR) and trends toward longer overall survival (OS), and progression-free survival (PFS) to atezolizumab in NSCLC, indicating its potential as an ICI biomarker [32].
2. Tumor dynamics monitoring during treatment
As plasma ctDNA concentration is largely determined by tumor burden, monitoring ctDNA level during treatment for patients with solid cancers can provide quantitative information on the changes in tumor burden over time. Contrary to clinical diagnostics such as computed tomography (CT), ctDNA detects tumor-specific variants and possesses a broad dynamic range [33]. Also, its half-life is shorter (from several minutes to 1 to 2 hours) than that of tumor markers [10]. In addition, ctDNA monitoring is noninvasive with a simple blood draw, and frequent monitoring is possible with repeated blood sampling. Thus, ctDNA is ideally suited for noninvasive, sensitive, real-time monitoring of tumor dynamics.
In numerous clinical studies across tumor types, the initial decrease in ctDNA level strongly correlates with clinical or radiographic response in patients undergoing treatment. In gastrointestinal and pancreatic cancers, a significant decrease in ctDNA levels prior to response evaluation correlated with CT response, indicating that an early change in ctDNA to chemotherapy predicted the later radiological response [19,23,34–37]. Early changes in ctDNA during treatment for NSCLC and breast cancer prior to radiographic evaluation correlate with ORR and significantly longer survivals [33,38–41]. Plasma ctDNA levels changed more rapidly and sensitively, and a positive correlation with PFS and OS was evident in many studies. In monitoring cancer treatment with ctDNA, it should be noted that a subset of patients may exhibit a transient peak in ctDNA levels 1–3 days after the start of radiotherapy or chemotherapy [34,37,42]. This transient peak is theorized to be the result of a brief period of rapid cell death during the earliest phase of treatment. However, the very early increase in ctDNA levels was ultimately indicative of treatment response in the patients and thus should be interpreted with caution [43]. Collectively, these findings suggest that “molecular response” revealed by ctDNA could serve as an early indicator of response prior to clinical manifestations and guide subsequent treatment. It should be noted, however, that although ctDNA monitoring is a good predictor of treatment response and prognosis, its utility in improving patient outcomes by ctDNA monitoring-based clinical decisions is uncertain, and prospective trials are required to answer this question.
In addition to its role as an early indicator of treatment efficacy, ctDNA monitoring can predict the emergence of acquired resistance and tumor progression much earlier than conventional clinical modalities. In patients with metastatic CRC treated with an anti-EGFR monoclonal antibody, treatment-emergent, subclonal alterations in KRAS, MET, and EGFR extracellular domain (ECD) causing acquired resistance to anti-EGFR were detectable in plasma as early as 10 months prior to the clinical tumor progression [22,44,45]. In CRC, breast cancer, and NSCLC treated with chemotherapy, longitudinal monitoring of ctDNA was able to detect molecular progression with a median molecular lead time of 1.3–3.3 months [36,46,47]. Whether “molecular progression” revealed by ctDNA monitoring has a true clinical significance, however, can only be determined by demonstrating that early treatment modification based on ctDNA results (e.g., early introduction of a subsequent line of treatment or therapy targeting somatic alterations identified by ctDNA) improves long-term survival or quality of life. Although this has not been demonstrated for the majority of clinical situations with molecular progression and warrants future randomized trials, the recently reported results from a randomized phase 3 PADA-1 trial are very intriguing because they provide a positive answer to the question posed above [48]. When estrogen receptor (ER)+ HER2− breast cancer patients with rising ESR1 mutation in real-time ctDNA monitoring during aromatase inhibitor (AI) therapy were randomized to fulvestrant+palbociclib vs. continuing AI+palbociclib, early introduction of fulvestrant and palbociclib therapy targeting ESR1 mutation based on ctDNA results improved PFS (median, 11.9 vs. 5.7 months; hazard ratio, 0.61), although OS has not yet been reported [48].
Noninvasive, real-time monitoring of ctDNA may be particularly important for immunotherapy patients, including those receiving ICI. It is well known that the response pattern to immunotherapy differs from that of other therapies, such as chemotherapy or targeted therapy. Immunotherapy may take longer to elicit a tumor response and it can be difficult to distinguish between real progression and pseudoprogression due to immune infiltration, especially early in the treatment [49]. It has been shown that the early change in ctDNA levels and on-treatment dynamics correlate strongly with PFS and OS in patients with ICI [50–52]. Therefore, real-time monitoring of ctDNA dynamics during treatment can identify patients who will benefit from immunotherapy and inform treatment decisions. In addition, ctDNA monitoring can help differentiate between true progression and pseudoprogression when tumor size increases due to immune infiltration before shrinking. ctDNA kinetics could accurately identify pseudoprogression in melanoma patients receiving ICI [53].
3. Molecular profiling of mechanisms for acquired resistance
Almost all patients with advanced solid cancers eventually experience tumor progression following the acquisition of novel alterations or the expansion of preexisting resistant clones during treatment. Molecular profiling of tumors at the time of acquired resistance has become increasingly important, especially with the advancement of novel treatment options targeting various alterations and the possibility of clinical trial enrollment. Mutational profiling with plasma ctDNA sequencing is particularly relevant at the time of tumor progression and acquired resistance because molecular profiling to identify mechanisms involved in acquired resistance requires the interrogation of the exact tumor that has progressed during treatment to capture the temporal changes in the tumor, but it is often difficult to biopsy the progressing tumor sites for sequencing in many cases. As ctDNA assays are noninvasive and repeatable, tumor profiling with ctDNA might be more desirable than tissue analysis. In addition, while acquired resistance may be caused by the emergence of multiple subclonal mutations from various metastatic tumors within the body, it is difficult for a tumor biopsy of a single metastatic site to capture spatial tumor heterogeneity and a comprehensive portrait of the molecular alterations involved in the development of acquired resistance. Indeed, polyclonal resistance alterations were common after anti-EGFR therapy, with 21% exhibiting ≥ 10 alterations [54], and EGFR ECD mutations were found to be highly heterogeneous (91% with a median of 4) in a separate study [55]. In a prospective study of 42 patients with gastrointestinal cancer and acquired resistance to targeted therapy, post-progression ctDNA identified clinically relevant resistance alterations and multiple resistance mechanisms more often than tissue biopsy, and 78% of patients had resistance alterations missed in the matched tumor [56]. Furthermore, during a screening for anti-HER2 therapy, some patients who tested negative for HER2 in tissue were found to be positive in ctDNA sequencing. This was attributed to the fact that the tissue samples had been collected before anti-EGFR therapy and did not show the current HER2 status of the tumors. These findings indicate that ctDNA sequencing could be better for tracking tumor changes after treatment and guiding further therapy for patients [29].
The time of tumor progression and acquired resistance is, therefore, the clinical scenario in which plasma ctDNA sequencing has the most clinically relevant and significant role in guiding treatment, at least in current clinical practice. The strong clinical basis of plasma ctDNA sequencing to identify acquired resistance at disease progression is reflected by the fact that the majority of guidelines endorse plasma ctDNA sequencing as a reasonable alternative or even a preference over tissue testing in patients with sufficient tissue for sequencing at disease progression.
According to National Comprehensive Cancer Network (NCCN) guidelines, plasma ctDNA sequencing using broad molecular profiling for genomic resistance mechanisms can replace tissue-based testing for NSCLC at tumor progression [57]. Particularly in EGFR mutant NSCLC that has progressed on a first- or second-generation anti-EGFR tyrosine kinase inhibitor, plasma ctDNA sequencing is recommended as an alternative to tissue-based T790M testing in clinical practice [4,8,57]. However, for maximum sensitivity, a reflex tissue testing is preferable for negative plasma test results.
Plasma ctDNA sequencing to identify PIK3CA mutation can be performed on ctDNA instead of tissue in hormone receptor–positive/HER2-negative patients following progression on or after an endocrine therapy in breast cancer to select patients for alpelisib plus fulvestrant according to NCCN guidelines [58]. Additionally, for ER-positive/HER2-negative breast cancer patients with progression on prior endocrine therapy, assays to identify ESR1 mutation should be tested preferentially in plasma ctDNA [4,58].
Furthermore, plasma ctDNA sequencing at the time of tumor progression and acquired resistance can also provide quantitative information on tumor dynamics that could be used to select a subsequent therapy, in addition to the qualitative aspects of novel alteration discovery. As treatment-emergent subclonal KRAS or EGFR ECD mutations or MET amplification decline after anti-EGFR therapy withdrawal, metastatic CRC could be re-sensitized to anti-EGFR agent rechallenge after an intervening interval [22]. While anti-EGFR rechallenge in CRC patients, selected only based on clinical criteria (interval between anti-EGFR therapies of 4 months), demonstrated a promising ORR (21%) [59], a pati-ent selection strategy for anti-EGFR rechallenge based on plasma ctDNA demonstrated an ORR of 30% and suggested it might be better for patients selection in a prospective study [60]. Multiple ongoing clinical trials are evaluating the clinical validity of ctDNA-guided anti-EGFR re-treatment in metastatic CRC patients who have been previously treated with anti-EGFR therapy [61–63].
Clinical Application of Plasma ctDNA Sequ-encing in the Management of Localized Solid Cancers
1. Minimal residual disease evaluation in the postoperative period
The extent of microscopic residual disease (minimal or molecular residual disease, MRD) in surgical fields or distant sites is believed to be directly related to the postoperative risk of recurrence in localized resectable solid cancers. The problem is that MRD is below the detection threshold of conventional diagnostic tools, such as CT scans. Hence, clinicopathologic characteristics, including pathologic staging, have been used as surrogate markers of recurrence risk, and adjuvant treatment is decided based on these characteristics. However, recent studies have demonstrated that the postoperative assessment of MRD with plasma ctDNA can identify patient at higher risk of recurrence better than clinical staging in a number of cancer types, including colorectal, breast, lung, urothelial and pancreatic cancers [64–77]. This indicates that detection of plasma ctDNA shed by MRD may be able guide postoperative adjuvant treatment strategy in terms of dose, intensity, and duration. A recently published randomized phase II DYNAMIC trial is the first study demonstrating the feasibility and clinical utility of the plasma ctDNA-guided adjuvant chemotherapy strategy [78]. The ctDNA-based decision of adjuvant treatment did not adversely affect patient outcomes (2-year recurrence-free survival of 93.5% vs. 92.4%) in stage II colon cancer while reducing the number of patients receiving adjuvant chemotherapy (15% vs. 28%) compared to SoC. This trial successfully examined the impact of a ctDNA-based adjuvant treatment decision strategy compared to a standard adjuvant decision in stage II colon cancer, and many ongoing clinical trials are investigating whether adjuvant chemotherapy can be modified based on postoperative ctDNA MRD status in various types of early-stage solid cancers [79–85] (Table 1). The prospective randomized trials of MRD-guided adjuvant treatment can be roughly divided into those with a superiority design of escalation strategy, where intensified treatment regimens are compared with SoC in patients with positive postoperative MRD, and those with a non-inferiority design of de-escalation strategy, where de-intensified regimens (e.g., shortened treatment duration, or reduced dose or even treatment omission) are compared with SoC in patients with negative postoperative MRD.
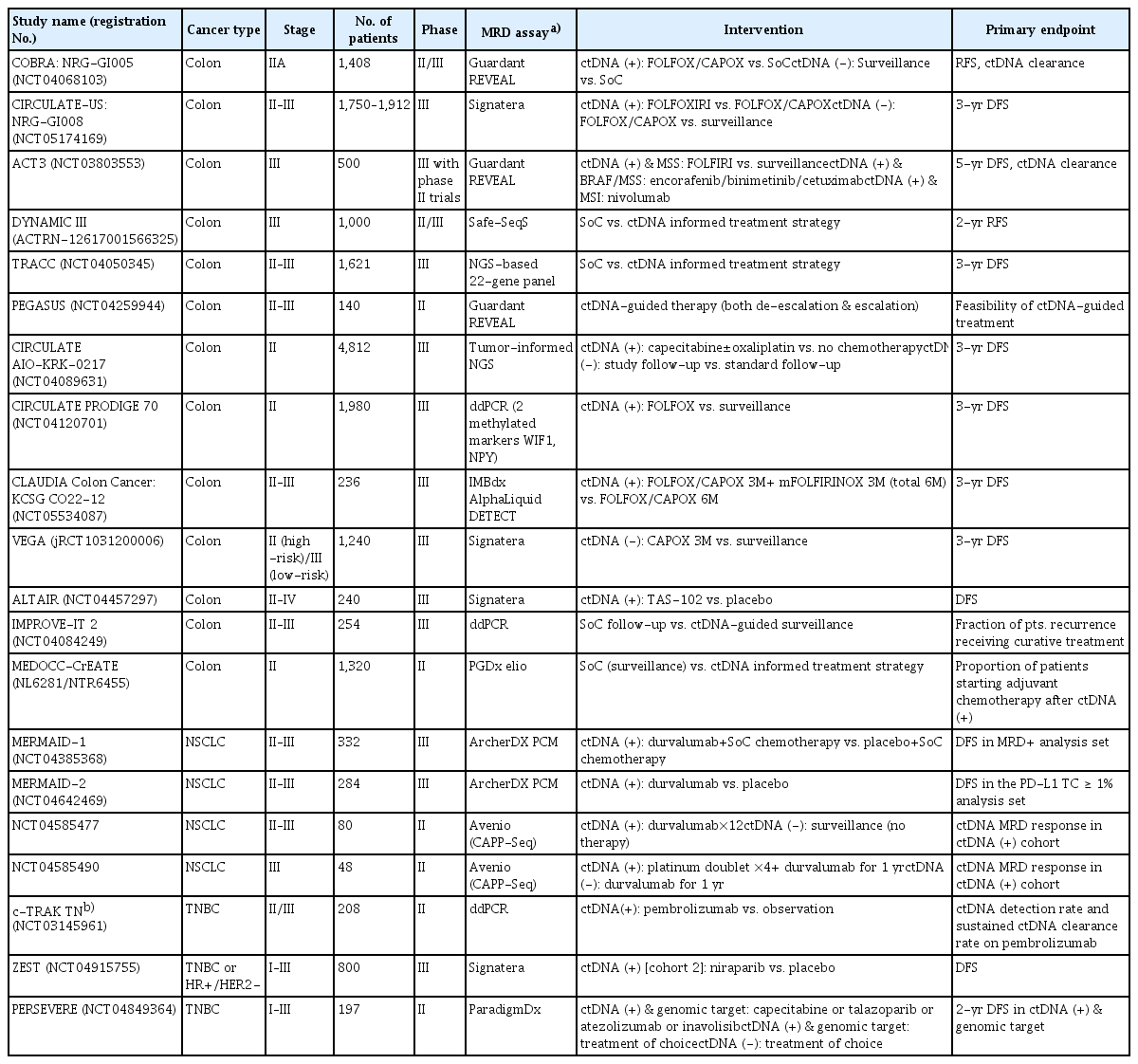
Representative MRD-directed adjuvant therapy trials in solid cancers
In addition, ctDNA-based MRD testing in the postoperative setting has the potential to evaluate the efficacy of adjuvant treatment and to direct treatment modification based on the information gathered. Because there is no macroscopic residual tumor, the effectiveness and benefit of adjuvant chemotherapy in cancer patients can only be evaluated partially during surveillance based on the occurrence of recurrence. In this regard, serial measurement of MRD dynamics during and after adjuvant therapy may permit quantitative evaluation of the efficacy of adjuvant therapy. Consequently, in patients with a rising ctDNA level during chemotherapy, the treatment can be regarded as ineffective and discontinued and alternative regimens can be implemented early before clinical recurrence.
Lastly, ctDNA-based MRD monitoring in the postoperative setting can be used to detect recurrence before clinical diagnosis of recurrence when tumor burden is below the detection thresholds of conventional modalities (“molecular recurrence”). However, even if plasma ctDNA-based MRD testing could detect molecular recurrence prior to overt recurrence, improvement of the long-term outcome by early treatment initiation at the time of molecular recurrence relative to conventional treatment initiation at the time of clinical recurrence must be demonstrated in clinical trials.
2. Treatment monitoring during neoadjuvant treatment
Currently, the response to neoadjuvant therapy (NAT) is assessed radiographically and/or pathologically in a variety of cancers, including lung, breast, ovarian, and rectal cancers, where NAT is among the SoC. Evaluation of response with clinical or pathologic tools is semiquantitative and difficult to perform frequently, particularly for pathologic response. In addition, clinical imaging is challenging to evaluate when the tumor burden is low. In contrast, residual tumor quantification using ctDNA has a greater dynamic range, is easily repeatable, and may be more sensitive for low tumor burden. In this regard, monitoring tumor dynamics with ctDNA during and after NAT has great potential because it could guide post-NAT management in several ways, including escalation/de-escalation of NAT or adjuvant therapy; delaying or omitting surgery (“watch-and-wait” or non-operative management [NOM]). In fact, positive ctDNA MRD during or after NAT was associated with an increased recurrence risk in locally advanced breast, rectal, gastric/gastroesophageal, and esophageal cancer, as well as in resectable CRC liver metastases, and even outperformed imaging in a few studies [86–100]. Recent randomized CheckMate-816 and I-SPY 2 are representative studies demonstrating the potential utility of ctDNA in NAT [90,101]. In light of these findings, novel clinical trials testing ctDNA-guided post-NAT management strategies can be envisioned, despite the infancy of ctDNA-directed post-NAT management. Considering that NOM is increasingly being investigated in early-stage cancers, such as rectal, breast, and esophageal cancer, ctDNA MRD could be utilized to identify patients who would benefit from NOM after NAT.
Multi-cancer Early Detection in the Preclinical Period
The Korean National Cancer Screening Guidelines curren-tly recommend early detection screening for patients at risk for gastric, colorectal, liver, breast, cervical, and lung cancers [102]. Although national guidelines for cancer screening methods and indications vary, the majority of cancers are not indicated for population-based screening, and most cases are diagnosed at a late stage [103]. Therefore, there is a significant need for early detection of various types of cancer, when cures and improved treatment outcomes are more likely to occur. Multiple features, including genomic, epigenomic, and proteomic markers, have been applied to ctDNA as a promising method for early detection of multiple cancers (MCED) [104–109]. While sensitivity has been the primary performance metric for screening tests, very high specificity is considered a prerequisite for the use of MCED tests in populations with a low incidence of cancer due to concerns about a high false-positive rate and overdiagnosis [108,110]. In large-scale validation studies with case-control cohorts, such as DETECT-A and Circulating Cell-Free Genome Atlas (CCGA), MCED tests demonstrated extremely high specificity with the false-positive rate of 1% and sensitivity ranging between 52% and 62% [104,107]. Despite the fact that their reported sensitivities fall short of standard screening tests for a single cancer, which range from 80% to 90%, ctDNA-based MCED tests could benefit the majority of cancers for which there is no standard screening test. Moreover, even in cancers with standard screening tools, when MCED is combined with standard screening tests, it could detect more patients with cancer and, more importantly, reduce the high false-positive rate, which is one of the weaknesses of standard screening [111,112]. However, while the true impact of ctDNA-based MCED tests can only be evaluated prospectively in the population they target, the majority of the reported sensitivity was derived from retrospective case-control cohorts, not the true target population. Therefore, the results of ongoing prospective studies, such as STRIVE (NCT03085888), SUMMIT (NCT03934866), PATHFINDER (NCT04241796), PREDICT (NCT04383353), and PRESCIENT (NCT04822792) are eagerly awaited.
Application of Plasma ctDNA Sequencing in the Clinical Trial Conduct and Drug Development for Solid Cancers
ctDNA has the potential to facilitate clinical trial conduct and early drug development in multiple ways beyond its role in clinical practice.
First, it can improve patient selection and enrollment in clinical trials of molecularly targeted drugs. Previously, a tissue-based assay was required for target enrichment in precision oncology clinical trial designs such as basket or umbrella trials. ctDNA-based target discovery can identify more patients with study-relevant targets than tissue analysis alone. In SCRUM-Japan studies, ctDNA sequencing-based clinical trial enrollment increased the trial enrollment rate (9.5% vs. 4.1%) and decreased the screening duration (11 vs. 33 days) in patients with gastrointestinal cancer in comparison to tumor tissue-guided trial registration [19]. It was also demonstrated that genetic alterations detected by ctDNA analysis effectively select patients for colorectal, gastric, breast, and pediatric cancer clinical trials [20,29,113–115]. In addition, ctDNA facilitates the discovery of tumor evolution leading to acquired resistance and the testing of drugs with potentially active resistance mechanisms [116].
Second, monitoring the dynamics of ctDNA during treatment could serve as an early and reliable surrogate endpoint of treatment efficacy [117,118]. In neoadjuvant and palliative settings, ctDNA changes over treatment may offer advantages over conventional endpoints, such as response, by providing a greater dynamic range, higher sensitivity, and easier temporal monitoring [39]. In an adjuvant setting, ctDNA positive conversion and clearance could also be used as early surrogate endpoints [119]. However, the use of ctDNA dynamics as a surrogate endpoint is exploratory at this time, and regulatory agencies require sufficient clinical data for approval. Future clinical trials should include ctDNA kinetics as endpoints and compare them with conventional endpoints.
Third, in addition to MRD-based treatment assignment in clinical trials, more clinical trials are using MRD-based patient enrollment in clinical trials because it can stratify patients into distinct risk groups and permit more efficient trial design and conduct [119,120]. In the recent Imvigor010 trial of adjuvant atezolizumab in urothelial carcinoma, ctDNA MRD status was shown to be a useful biomarker for identifying patients who will benefit from the treatment [72].
Fourth, the potential of ctDNA-guided clinical trials may be more promising in immunotherapy clinical trials [121,122]. As clinical response to immunotherapy may differ from conventional therapy in terms of response timing and pseudoprogression, relying on conventional methods to evaluate tumor response may be misleading.
Practical Considerations for the Application of ctDNA in Solid Cancers
The amount of ctDNA in plasma (ctDNA fraction) is significantly influenced by clinical factors that affect tumor shedding and preanalytical factors, such as blood collection, storage, and DNA isolation, that have a significant impact on its sensitivity. Consequently, the decision regarding ctDNA testing and the interpretation of its results should be made with sufficient knowledge of the various factors that may influence the results. While preanalytical factors are indeed important for ctDNA analysis, we will focus solely on clinical parameters in this review, as preanalytical factors are beyond its scope.
Although the overall sensitivity for ctDNA detection was 73.5% among > 10,000 Chinese patients with cancers and > 70% for most stage IV samples, some cancer types, including renal cancer, melanoma, and brain tumors were among the least shedding cancers with relatively low detectability [11,123]. Accordingly, caution should be exercised in interpreting ctDNA testing results for these non-shedding tumors, and negative results should be confirmed by tissue assays. As shedding of ctDNA is known to be active in growing tumors [124], whereas stable tumors are unlikely to shed enough ctDNA to be detected, blood sampling for plasma ctDNA sequencing should be preferentially performed in growing tumors. Furthermore, CRC patients who do not have liver metastases are found to have significantly lower levels of ctDNA than those with liver metastases, which can lead to reduced sensitivity for detecting subclonal variants and lower concordance rates between ctDNA and tissue [125,126]. Hence, the negative results with metastatic CRC without liver metastases may be a result of low ctDNA levels in the blood, and interpretation should be made with caution.
Similarly, patients undergoing treatment (chemotherapy or radiotherapy) or who have recently completed treatment are unlikely to shed ctDNA; therefore, ctDNA analysis should be performed with caution on these patients. After surgery and intervention, normal DNA is released into the blood, diluting and decreasing ctDNA sensitivity. This could be a significant problem for qualitative monitoring or MRD testing using ctDNA. In a study of patients with colorectal and bladder cancer, it was found that levels of total cfDNA increased for up to 4 weeks following surgery [127]. While the optimal timing of blood sampling for MRD assay needs to be determined in future studies considering the type and invasiveness of the surgery and the sensitivity of individual assay, it would be safer to use blood samples collected at least 3 weeks after surgery to minimize the false-negative issue.
Since many clinical ctDNA sequencing panels do not include sequencing of peripheral blood mononuclear cells (PBMCs), mutations in cancer susceptibility genes such as BRCA1/2 or PALB2 detected in ctDNA results may be germline rather than somatic in origin. Therefore, germline testing should be considered when mutations are found in plasma for these genes. Clonal hematopoiesis (CH) is the clonal expansion of hematopoietic stem cells that increases with age and is usually in low fractions (less than 1%) of plasma, which can be mistaken for somatic mutations. A study that used parallel sequencing of plasma and matched PBMCs found that 14% of samples from patients with cancer contained CH variants [11]. Given that several genes, including 15 canonical genes (DNMT3A, TET2, ASXL1, PPM1D, TP53, JAK2, RUNX1, SF3B1, SRSF2, IDH1, IDH2, U2AF1, CBL, ATM, and CHEK2) are well-known for association with CH [128], interpretation of ctDNA results should be cautious when low-frequency variants in these genes are found in plasma, particularly in elderly patients. If the results could affect treatment decisions, additional tumor assay or germline testing should be considered.
Future Directions and Perspectives
Plasma ctDNA sequencing is being rapidly incorporated into clinical practice for tumor profiling in the management of solid cancers, while its application in other areas, such as MRD detection and monitoring, or MCED is being actively investigated in a large number of ongoing clinical trials. Table 2 summarizes the currently recommended clinical indications for plasma ctDNA sequencing in the management of solid cancers. Notably, the current clinical application of plasma ctDNA sequencing is limited to a qualitative evaluation and remains complementary to tissue-based assays in the majority of indications.
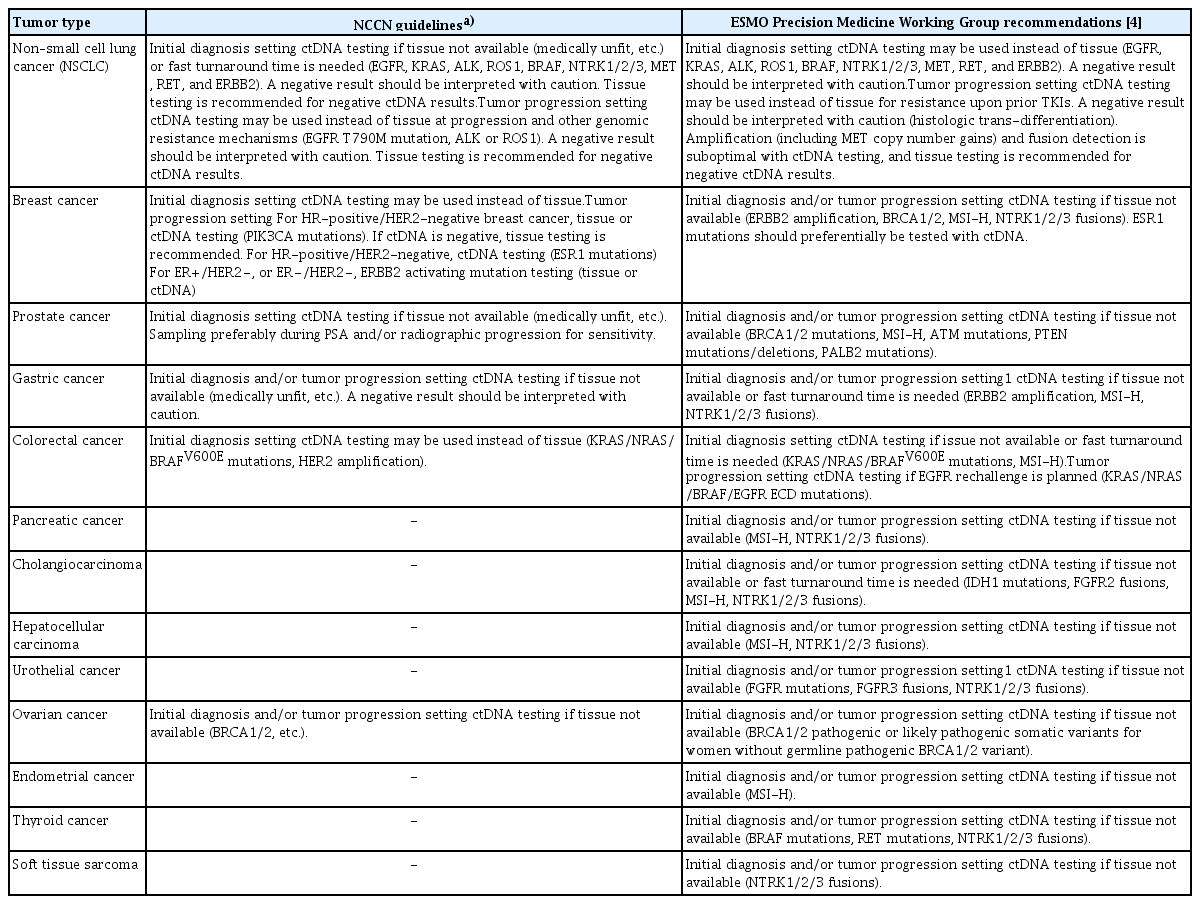
Currently recommended clinical indications for plasma ctDNA sequencing in solid cancers
In the future, plasma ctDNA sequencing may achieve an accuracy equal to or even greater than that of the tissue-based assay if increasing coverage depth, decreasing NGS costs, and the introduction of newer technologies can compensate for the low concentration [129]. As plasma ctDNA has many practical advantages over tissue testing, including ease of sampling, rapid TAT, repeatability, and potential to overcome spatial heterogeneity, the ongoing technical development of ctDNA sequencing foresees its future application in a much broader range of solid cancer management settings.
In addition to its expanding clinical applications coupled with improving accuracy, plasma ctDNA is being studied within the context of comprehensive multi-omics, such as WGS, methylation profiling, nucleosome mapping, and fra-gmentomics, as well as machine learning algorithms to uncover biological features from complex datasets, highlighting its potential to provide deeper insights into cancer biology [17,130].
Notes
Author Contributions
Conceived and designed the analysis: Cha Y, Han SW.
Wrote the paper: Cha Y, Kim S, Han SW.
Conflicts of Interest
Yongjun Cha received a consulting fee from IMBdx, Inc. Sheehyun Kim disclosed no conflicts of interest. Sae-Won Han received research funding support (institutional) and honoraria from IMBdx, Inc.
Acknowledgments
This work was supported by grants from the National R&D Program for Cancer Control (HA22C0062) through the National Cancer Center funded by the Ministry of Health & Welfare, Republic of Korea and from Patient-Centered Clinical Research Coordinating Center (PACEN) (HC21C0149) funded by the Ministry of Health & Welfare, Republic of Korea.