Reclassification of Five BRCA1/2 Variants with Unknown Significance Using Complex Functional Study
Article information

Abstract
Purpose
While BRCA1/2 genes are commonly investigated, variants of unknown significance (VUS) and variants with potential splice effect are still being detected and they represent a substantial challenge in genetic counseling and therapy.
Materials and Methods
Out of genetically tested 3,568 hereditary breast and ovarian cancer probands five, functionally not investigated variants with potential splice-modifying effect were subjected to functional characterization. Transcript-level analysis on peripheral blood-derived RNA of the carriers was performed to test aberrant splicing. The completeness of the aberrant splicing event was also studied, existence and extent of nonsense-mediated decay was even addressed. Clinical and phenotype data, pedigree and co-segregation analyses were also done. Locus-specific loss of heterozygosity (LOH) in tumor tissues was additionally tested.
Results
In case of the BRCA1:c.4484+4dupA and the BRCA1:c.5407-10G>A variants functional results allowed us to reclassify them from VUS into likely pathogenic category. BRCA1:c.4358-31A>C, by producing incomplete aberrant splicing, was highlighted as strong VUS, but in lack of other supporting evidence, re-categorization was not possible. The likely pathogenic assertion of previously not reported BRCA2:c.8487G>T was reinforced based on its spliceogenic property and tumor LOH, while BRCA2:c.793G>A failed to present aberrant splicing in spite of suggestive predictions, which altered its original VUS evaluation into likely benign class.
Conclusion
We presented molecular and clinical evidence for reclassification of four out of five BRCA1/2 variants. Both up- and down-classification harbour important clinical significance. Patients carrying re-classified pathogenic variants in the future will not be dropped out from medical surveillance, preventive measures, treatment and predictive family screening in relatives at risk.
Introduction
As high-throughput next-generation sequencing (NGS) is becoming more and more recognized in routine diagnostics, there are an increasing number of novel rare variants, which are either not registered in locus-specific databases or clinically not interpreted. These variants with uncertain significance (VUS) pose challenge to genetic counseling and clinical managements [1,2]. Regarding BRCA1/2 genes, it is recommended to report VUS in the clinical genetics test records by the European consensus statement and expert recommendations [3]. However, VUS should not be used for medical decisions (surveillance, treatment, or preventive measures) or for predictive testing in relatives at risk; therefore, patients harboring such genetic alterations cannot benefit from the mutation-based therapies. This explains the strong demand to assert these variants into definite pathogenic or benign clinical categories aided by various gene-based functional studies. The American College of Medical Genetics and Genomics (ACMG) elaborated the standards and guidelines for the interpretation of sequence variants through the synthesis of categorical evidence [4]. According to these guidelines, well-established functional studies, for example, mRNA-level tests examining these variants’ possible adverse effects on splicing may promote their pathological assertion. In the further assessment of the clinical relevance of VUS, variant-phenotype co-segregation in the family by clinical geneticists, potential loss-of-heterozygosity testing or functional in vitro assays represent important landmarks [2,4].
Germline pathogenic variants of the BRCA1 and BRCA2 genes account for 15%–20% of hereditary breast and ovarian cancer (HBOC) cases and represent the main genetic cause of hereditary familiar tumors of these types [5]. The Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Consortium [6] registers and curates BRCA1/2 variants in the BRCA Exchange database [7]. The consortium assembles genetic and clinical information originating from international expert laboratories in order to categorize these variants based on gene-specifically calibrated criteria (ver. 2.5.1, 29 June 2017). Another valuable repository for annotated BRCA1/2 variants is the LOVD (Leyden Open Variation Database, https://grenada.lumc.nl/LSDB_list/lsdbs), especially that, curated by HCI/Tavtigian (http://hci-exlovd.hci.utah.edu/home.php). Still, a substantial amount of BRCA1/2 variants fall into the VUS category, approximately 5%–10% of patients who undergo genetic testing of BRCA1 and BRCA2 receive a result reporting a VUS [8]. The most VUS are extremely rare or not even registered in population databases. Our department has performed routine genetic testing of HBOC families for germline mutations of BRCA1/2 genes with NGS techniques for over 6 years. During this period, we tested 3,568 probands, whose personal and/or family history of tumors conformed to the criteria of the relevant National Comprehensive Cancer Network (NCCN) guidelines for genetic testing [9]. In the course of the genetic diagnostic workflow, we regularly detected variants, which were either clinically not reviewed in the proper locus-specific databases, or their functional assessment was conflicting, so we regarded them as VUS. Besides VUS, variant classification is a dynamic process, and previously classified variants sometimes need periodic reevaluation. The knowledge base for variant classification is continuously increasing with the expanding data in both public and in-house databases, publications reporting functional studies, as well as improvements in computational algorithms for predicting pathogenicity and genotype-phenotype association [10]. Therefore, following current recommendations, our laboratory periodically reviews previously identified genetic test results and performs variant-level reassessment.
The relevance of splicing in the BRCA genes, whether alternative or aberrant, was reported in various studies in connection with functionality [11,12]. Since any type of genetic variation (missense, nonsense, synonymous as well as intronic) may influence correct splicing, we systematically subjected these variants to diverse in silico splice predictions, and those that were predicted to be potentially spliceogenic, were further analyzed. We selected five VUS with potential splice effect and we studied them at transcriptional level, using blood RNA samples of the proband. Additional clinical evidence, as clinico-pathological features of carriers, pedigree analysis during clinical genetic counseling, presence of potential locus-specific loss of heterozygosity (LOH) in the corresponding tumor tissues and co-segregation of the variant with the disease were involved in the establishment of the plausible clinical relevance.
We present transcript-level genetical evidence as well as phenotype rationales for reclassification of five BRCA1/2 variants.
Materials and Methods
1. Patients, clinical genetic counseling, and BRCA1/2 genotyping
In this study, we analyzed 3,568 Hungarian HBOC patients by NGS method within the frame of routine genetic testing for BRCA1 and BRCA2 genes at the Department of Molecular Genetics of the National Institute of Oncology, Budapest, Hungary between 2015–2020. In Hungary, a national guideline was published in 2020 by the Board of Clinical Geneticists about the criteria for germline testing of patients with breast cancer (http://www.hbcs.hu/uploads/jogszabaly/3278/fajlok/2020_EuK_20_szam_EMMI_szakmai_iranyelv_2.pdf). In brief, breast/ovarian cancer under the age of 50, triple-negative breast cancer or ovarian cancer or male breast cancer at any age, breast cancer at any age with two or more first-degree relatives (1 ≤ 50) or at least one ovarian first-degree cancer relatives [13]. Prior to molecular genetic testing, clinical genetic counseling was performed in each case according to the Hungarian legal and ethical regulations, where personal and familial tumor history was registered. All participants gave written informed consent for the genetic testing. Genotyping was carried out for all coding exons and exon-intron boundaries of BRCA1 and BRCA2 genes with Multiplicom amplicon-based enrichment BRCA MASTR Dx or BRCA MASTR Plus Dx library preparation kit (Agilent Technologies, Santa Clara, CA) and sequenced on MiSeq Illumina platform (Illumina, San Diego, CA). Bioinformatics analysis was done with the MASTR Reporter software v.1.1 (Agilent Technologies). When a VUS was identified, the significance of this result was extensively explained and discussed with the patients during the post-test counseling. Then, if the patient agreed to participate in research studies (including in vitro characterization and family screening for studying segregation), a second sampling was performed and these samples were further used for functional characterization. Upon conclusive result, the patients were re-counseled in the light of the new result. The availability of genetic testing was offered for all at-risk relatives of the variant carriers’ families.
2. In silico predictions and variant selection
Splice alteration predictions for splice consensus regions (−3 to +8 at the 5′ splice site and −12 to +2 at the 3′ splice site) were taken from ADA_score and RF_score, arising from adaptive boosting [14] and random forest ensemble [15] learning methods integrated into the annotations of dbNSFP v4.0 [16]. Cutoff scores > 0.9 for ADA and > 0.7 for RF were considered. Possible splice effects of intronic variants outside of the consensus splice regions were queried by varSEAK, an online public access program (JSI Medical Systems, Ettenheim, Germany), based on mainly MaxEntScan calculations. Scores ≥ 4 were taken as a cutoff for plausible splice impact. RNABP (http://nsclbio.jbnu.ac.kr/tools/RNABP) [17] and LaBranchoR [18] predictors were applied for determining 3′ splice branchpoint positions. LaBranchoR defines the most probable position of the active adenine and RNABP predicts the odds for a nucleotide being a potential branchpoint site. Variants were selected for cDNA-level study if any of these predictions were suggestive of possible aberrant splicing and variant frequency was extremely low (< 0.001) in various populations. Variants, elected for transcript-level analysis are listed in Table 1. Phenotype and family characteristics of the probands carrying these variants are listed in Table 2 and S1 Fig.

Five variants selected for transcript analysis according to in silico predictions
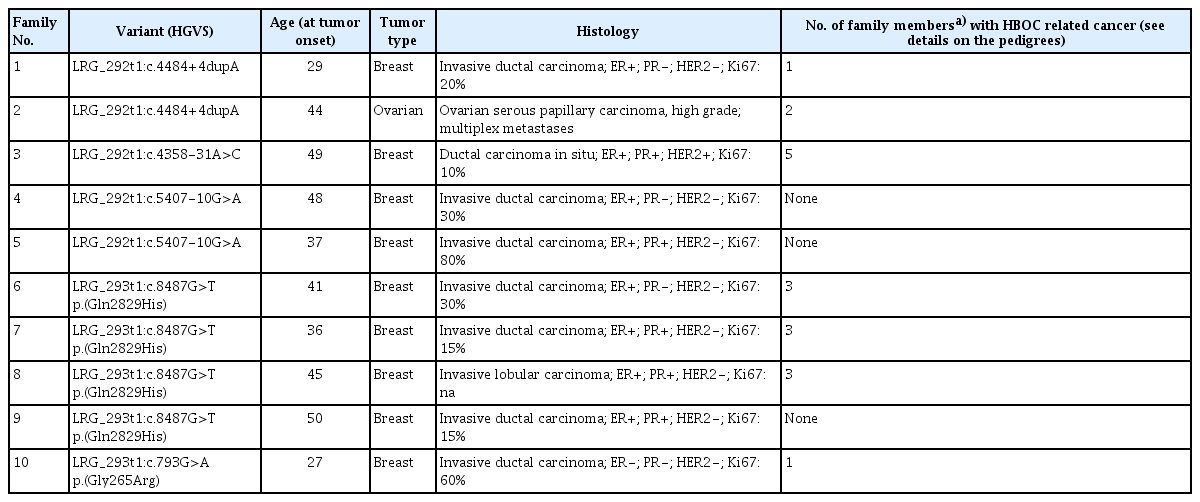
Baseline characteristics of index patients from each family
3. cDNA qualitative analyses
RNA was isolated using the Tempus Spin RNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA) from peripheral blood taken in Tempus Blood RNA tubes (Thermo Fisher Scientific) according to the manufacturer’s recommendations. First-strand reverse transcription was carried out by SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific). Reverse transcription PCRs (RT-PCRs) amplifying the variant-containing exons along with at least two adjacent exons were designed individually (list of cDNA primers is given in S2 Table). Amplification products were visualized on 1% agarose gel next to Hyper Ladder 1 kb DNA sizing standard (Bioline, London, UK) and subsequently sequenced by conventional Sanger sequencing method on ABI3130 Genetic Analyzer using the BigDye v.1.1 Kit (Thermo Fisher Scientific). Sequencing was done for the whole RT-PCR product without separation of the respective bands to compare peak intensities of normal and aberrantly spliced products. Where it was necessary to remove the interfering predominant normal alternative splice product, fragments of different sizes were cut out and cleaned from the gel by Monarch Gel Extraction Kit (New England Biolabs, Ipswich, MA) and the purified product was sequenced as above.
4. cDNA semi-quantitative measurements
Relative quantitation of the normal and aberrantly spliced isoforms was assessed by two analytical methods: quantitative real-time PCR (qPCR), as well as quantitative multiplex PCR of short fluorescent fragments (QMPSF), and the averaged results of the two methods were considered for the subsequent calculations. Selective amplification of the two types of transcripts was performed with specific primer pairs engineered to discriminate between the two splice forms. At least one primer of the pairs was designed so that it should span exon borders, to amplify only from the cDNA (exact primer sequences are listed in S2 Table). qPCR was run on QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) in relative quantification mode using SYBRGreen chemistry (Xceed HRM 2× Mix, Institute of Applied Biotechnologies, Prague, Czech Republic). Since both types of transcripts were amplified from the same template cDNA, no calibrator sample was needed, the expressions of the normal and aberrantly spliced products were directly comparable. Each test was performed in technical triplicates and three independent measurements were done. Means of the measurements with standard deviations were calculated. For QMPSF, one of the primer pairs of the respective amplicons was labeled with FAM fluorescence. PCR was conducted using Qiagen Multiplex PCR kit (Qiagen, Hilden, Germany) for 24 cycles at 62°C annealing temperature. The resulted products were subjected to capillary electrophoresis on an ABI3130 Genetic Analyzer (Thermo Fisher Scientific) in Microsatellite analysis mode. Peaks were visualized using the Peak Scanner Software 2.0 provided for the instrument. Peak ratios were calculated based on the area under the curve (AUC). Three biological measurement replicates were done.
Relative allelic expressions for allelic imbalance tests were measured on cDNA calculating the AUC ratios of exonic heterozygote positions in sequencing electropherograms [19]. The peak ratios defined on cDNA were normalized to the ratios of the same positions measured on gDNA.
5. LOH tests
Loss of the normal allele was tested in the tumor DNA of the probands, where it was available. DNA was extracted from the tumor using the Maxwell RSC DNA FFPE kit (Promega Corporation, Madison, WI). The PCR-amplicon of the variant-containing region was subjected to sequencing and allelic AUC ratio of the electropherogram peaks at the variant position was determined. LOH was calculated by normalizing the AUC ratios of the variant position of the tumor to that of the gDNA, and the tumor content was also taken into account by using the formula below:
6. Complex evaluation of pathogenicity
We employed the VarSome software’s built-in pathogenicity calculator [20], corresponding with the statements of Goldgar et al. (2004) [21], for allocating variants into the 5-tier categories along with current ACMG guidelines [4]. As additional supporting evidence, at some variants we took into consideration co-segregation, LOH, family history, and proband phenotype characteristics to underpin their clinical relevance.
Results
1. Selection of VUS potentially affecting splicing
In the course of our routine BRCA1/2 diagnostic NGS-sequencing, we tested altogether 3,568 probands whom clinical presentation fulfilled the HBOC criteria for genetic testing (S3 Fig.) [9]. As a result of the comprehensive exon and exon-intron boundary sequencing of both genes, we detected 560 different variants, 130 of which were VUS according to the relevant ACMG criteria or not registered in BRCA1/2 locus-specific databases. The majority of them were extremely rare or absent in various population databases. We subjected these variants to diverse in silico splice prediction algorithms defining canonical splice disruptions or creation of novel splice sites (see “Materials and Methods”). Seven of the variants were suggestive for having spliceogenic effect by at least one of the in silico tools and for five variants out of them (with pan-population frequencies < 0.01 each), RNA samples were available for transcript-level analysis (Table 1). The study involved 10 nonrelated families altogether, carrying any of these five variants (Table 2, S1 Fig.).
2. BRCA1 intronic variants causing partial exon 14 skipping
Two probands of nonrelated breast cancer families (family 1 and family 2) carried a BRCA1 c.4484+4dupA variant, which was an insertion of an additional adenine nucleotide after the 4th basis of the BRCA1 intron 14, close to the canonical splice donor site. VarSeak prediction gave a high score for splice alteration (Table 1). Another proband in a different family was a c.4358-31A>C variant carrier (family 3). Branchpoint predictors anticipated that this latter variant affects the active adenine upstream the splice acceptor site of exon 14 (Table 1, S4 Fig.). RT-PCR amplification from the cDNA of the variant carriers with primers flanking exon 14 yielded a smaller-sized extra band in all three cases (Fig. 1A), which was sequenced and identified as an aberrant splice product with whole exon 14 skipping (Δ14) (Fig. 1B, S5 Fig.).

BRCA1 c.4484+4dupA and BRCA1 c.4358-31A>C variants both cause BRCA1 exon 14 skipping (A–F). (A) RT-PCR results of probands of F1 and F2 (BRCA1 4484+4dupA carriers) and F3 (BRCA1 c.4358-31A>C carrier) yielded an additional 150 bp shorter product on agarose gel, not present in controls. (B) Sanger sequencing of the RT-PCR products of the variant carriers showed whole exon 14 skipping in all probands, with a lower extent in the case of F3. The sequencing shows the reverse direction. (C) Allelic imbalance test of a nearby heterozygote exonic variant BRCA1 c.4837A>G in F1 shows a 1:2 ratios for the A:G alleles on cDNA relative to gDNA. (D) Relative abundance of the FL (blue bars) and Δ14 (red bars) transcripts measured by qPCR in F1, F2, and F3. Error bars represent standard deviation of three measurements. (E) Relative abundance of the FL and Δ14 transcripts measured by QMPSF in BRCA1 4484+4dupA carrier (F1) and BRCA1 c.4358-31A>C carrier (F3). (F) Detection of incomplete aberrant splicing on RT-PCR product amplified exclusively from the FL transcript by sequencing tagging polymorphism BRCA1 c.4837A>G. The electropherogram of the variant position is enlarged on the right side. (G) The composition of FL and Δ14 transcripts from the wild type (blue) and variant carrier (red) alleles in BRCA1 4484+4dupA carrier (F1) and BRCA1 c.4358-31A>C carrier (F3). The green segment represents the NMD-degraded fraction. The extent of FL transcript originating from the BRCA1 c.4358-31A>C carrier allele is only imputed. FL, full length; F1, family 1; F2, family 2; F3, family 3; C1–3, wild type controls; L, 1 kb ladder (Promega); NMD, nonsense-mediated mRNA decay; RT-PCR, reverse transcription polymerase chain reaction; QMPSF, quantitative multiplex PCR of short fluorescent fragments; qPCR, quantitative real-time polymerase chain reaction.
Semi-quantitative real-time PCR, as well as QMPSF analyses designed for specific amplification of the normal and aberrantly spliced RNA products showed that the Δ14 transcript is present in a lower quantity than the normal, full-length transcript (Fig. 1D and E). The average proportions were 0.32 (approx. ratio 1:3) for c.4484+4dupA carriers and 0.17 (approx. ratio 1:5.5) for c.4358-31A>C carrier calculated based on both detection methods. In addition, one c.4484+4dupA carrier proband carried numerous exonic heterozygote variants that allowed testing of allelic imbalance. Interestingly, AUC calculations of a heterozygote position in exon 16 (c.4837A>G) showed a 1:2 ratio of the two nucleotides in the electropherogram superposition (Fig. 1C). To resolve the discrepancy between the two ratios (1:3 and 1:2), we raised the possibility that incomplete aberrant splicing may interfere with the allele expression ratios measured. We tested this hypothesis using the c.4837A>G variant as a tagging polymorphism: we performed allele-specific RT-PCR with primers amplifying only the full-length, exon 14-containing transcript encompassing the c.4837A>G variant position. Concomitant Sanger sequencing of the resulted PCR product and measurement of the AUC of the electropherogram peaks in the tagging position yielded that the ratio was A:G=1:5.5 (Fig. 1F). Regarding the fact that Δ14-containing allele-specific PCR resulted exclusively in allele A, we could state that allele A is in “cis” position with 4484+4dupA variant, therefore the presence of allele A in the full-length transcript with 1:5.5 ratio markers that ~20% of the 4484+4dupA variant-containing allele also produced normally spliced RNA product. A remarkable portion of the remaining ~80% aberrantly spliced product may be partially degraded by nonsense-mediated decay (NMD), leaving only ~40% of the allelic expression as aberrant transcript (Fig. 1G). This indirectly suggests that NMD degrades approximately half of the BRCA1 Δ14 transcript. Unfortunately, the two other probands did not carry any exonic alterations in heterozygote form. Consequently, the calculation of the measure of incomplete aberrant splicing was feasible only indirectly in the case of the c.4358-31A>C carrier. Calculating with the same extent of NMD (~50% degradation of the BRCA1 Δ14 transcript), we yielded that nearly half of the variant-carrier allele might be normally spliced. Therefore, most probably, only half of the amount of aberrant splice product was detectable at c.4358-31A>C carrier relative to c.4484+4dupA carrier (Fig. 1G), which may be attributable to the larger incompleteness of this variant’s aberrant splicing effect.
The pedigrees of the three families are depicted in S1 Fig. All showed characteristic personal and familial HBOC features (Table 2). Family members in family 1 were available for genetic testing allowing genotype-phenotype co-segregation analysis. The proband’s mother, who was nonaffected turned out to be a non-carrier. The paternal grandmother, who had breast cancer at the age of 54, carried the variant. The proband’s father turned out to be also a variant carrier, but without clinical symptoms.
3. BRCA1 c.5407-10G>A causes partial intron inclusion
The BRCA1 c.5407-10G>A variant was detected in two independent probands in our tested cohort (family 4 and family 5) and both harbored characteristic personal and familial HBOC features (Table 2, S1 Fig). The variant changes a G nucleotide to an A in intron 22, ten nucleotides ups-tream of the exon 23, which was predicted in silico to disturb canonical splicing (Table 1). RNA sample was available from the proband of family 4. RT-PCR amplification yielded a fragment, which was indistinguishable from the wild type in length. Nonetheless, sequencing analysis of the fragment revealed aberrant splicing with retention of eight nucleotides of intron 22 upstream of the BRCA1 exon 23 (Fig. 2A). The nucleotide change introduced a novel AG acceptor dinucleotide within the AG exclusion zone [22], which acted as a novel strong acceptor site. Since the mutant transcript generated stop codon only in the last exon (exon 24), the resulting aberrant transcript was not subject to NMD. This was verified on the cDNA by a heterozygote exonic position, which actually did not show allelic imbalance (Fig. 2B). Therefore, the relative abundance of the normal and alternative transcripts reflects reliably the original ratio of the two splicing events. This was also confirmed by QMPSF technique (Fig. 2C). The completeness of the aberrant splicing was also studied applying a tagging variant c.4837A>G in exon 16, which was present in heterozygote form in one of the carriers. The tagging variant was co-amplified in a specific PCR reaction, which was designed for selective amplification of the normal, wild-type transcript. Sequencing electropherogram of the tagging variant position yielded only the G allele, no traces of the A allele (which was “in cis” with c.5407-10G>A) was detectable (Fig. 2D). This result ascertained that all the transcripts generated from the c.5407-10G>A variant-carrier allele were aberrant, so the aberrant splicing induced by this variant was complete. Furthermore, LOH test of the breast tumor tissues of index cases was available both in family 4 and family 5. Loss of the normal allele was demonstrated in both cases with R=0.26 and R=0.3 scores, respectively (Fig. 2E).
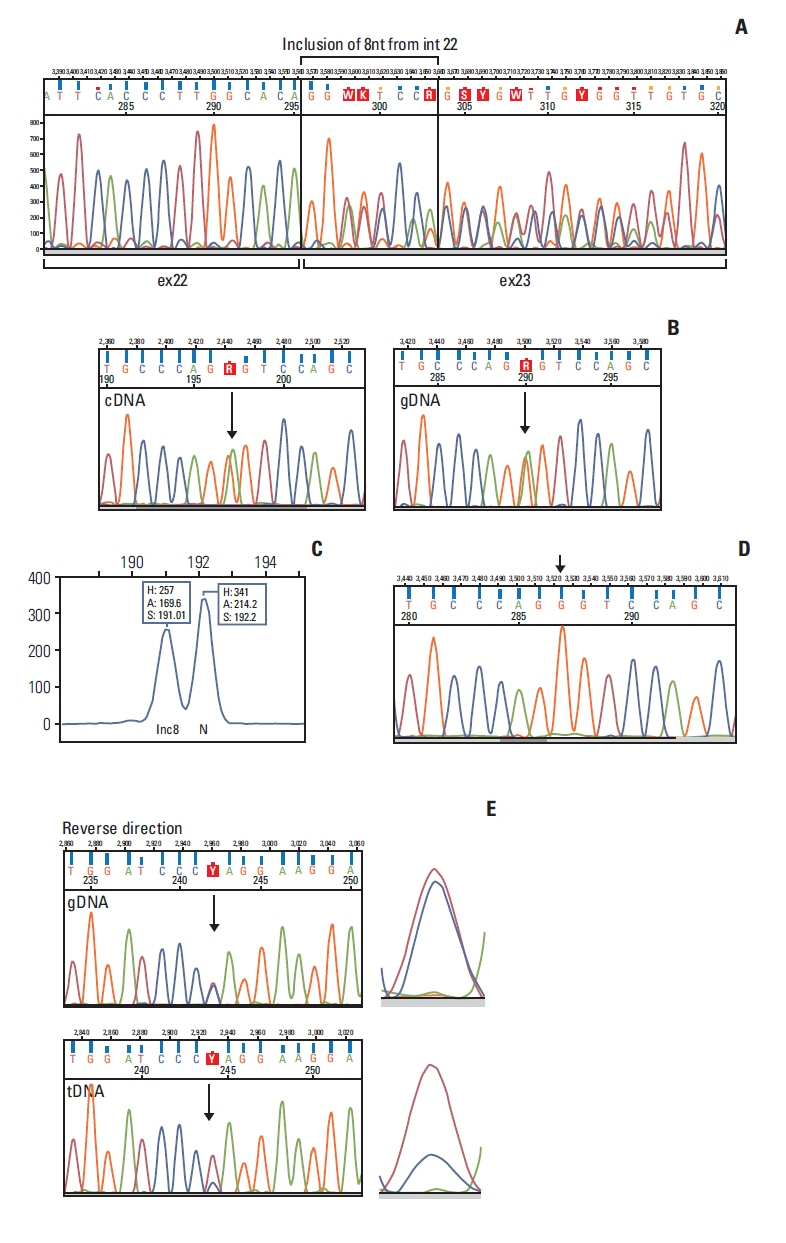
cDNA analysis of BRCA1 c.5407-10G>A variant. (A) Sanger sequencing result of the RT-PCR product of the BRCA1 c.5407-10G>A variant carrier proband of F5. Aberrant transcript revealed the inclusion of eight nucleotides of intron 22 into exon 23 generating a frameshift from this position. The peak intensities of the normal and aberrant sequences are equal. (B) Allelic imbalance test harnessing a heterozygote position BRCA1 c.4837A>G outside of the aberrantly spliced exon shows a 1:1 allelic ratio compared to the same position in gDNA. This confirms that the aberrant transcript is not degraded by NMD. (C) Relative abundance of the normal (N) and aberrant (inc8) transcripts measured by QMPSF. The slight difference may arise from suboptimal specificity of the discriminative primers. (D) Detection of complete aberrant splicing on RT-PCR product amplified exclusively from the normal transcript by sequencing tagging polymorphism BRCA1 c.4837A>G. Arrow points to the variant position, which represents only G, corresponding to the wild type allele. (E) Representative example for LOH in family 4. The electropherogram of the variant position is enlarged on the right side. F5, family 5; gDNA, genomic DNA; inc8, aberrant transcript with 8 nucleotide inclusion from intron 22; LOH, loss of heterozygosity; N, normal transcript; NMD, nonsense-mediated mRNA decay; QMPSF, quantitative multiplex PCR of short fluorescent fragments; RT-PCR, reverse transcription polymerase chain reaction; tDNA, tumor DNA.
4. Transcript-level study of BRCA2 putatively spliceogenic exonic variants
Based on in silico predictions, we selected two different BRCA2 variants for cDNA analysis, for which it was anticipated that the canonical splice sites might be affected. Of these, BRCA2 c.8487G>T, positioned in the last nucleotide of the BRCA2 exon 19, occurred in four unrelated probands in our cohort 4/3,568 (0.11%). Blood RNA sample was available from only one proband (family 6). RT-PCR–amplification of the region flanking the variant carrier exon yielded two products: one corresponded to the full-length transcript, while the other, shorter fragment proved to be an aberrant transcript with whole exon 19 skipping (Fig. 3A). The peak intensities of the sequencing electropherogram at the superposition of the normal and aberrant transcript sequences were equal (Fig. 3B); therefore, we suspected that the aberrant splicing was complete. Indeed, a tagging exonic variant position (c.7242A>G), which was also present in the proband in heterozygote state, we detected only the allele G, when amplifying the normal transcript selectively (Fig. 3C). No traces of allele A was present, implying that no full-length transcript was generated from the c.8487G>T variant carrier allele. Tumor sample DNA was available from three probands (family 6, 7, 9). LOH was demonstrated in all three samples with a mean Z score=0.25 (standard deviation, 0.03) (Fig. 3D).

cDNA analysis of BRCA2 exonic variants. Panels A–D show the results of the BRCA2 c.8487G>T variant. (A) RT-PCR gel electrophoresis results of proband of F8 (BRCA2 c.8487G>T carrier) yielded an additional 120 bp shorter product on agarose gel, not present in controls. (B) Sanger sequencing of the RT-PCR products of the variant carrier showed whole exon 19 skipping. Reverse direction sequencing is depicted. (C) Demonstration of complete aberrant splicing on RT-PCR product amplified exclusively from the FL transcript. At the tagging BRCA2 c.7242A>G variant position only G allele was detected and not any A (arrow points it out). (D) Representative example for LOH in family 7 highlighting the heterozygote superposition of the electropherogram in gDNA versus tDNA. (E, F) The results of the BRCA2 c.793G>A variant. (E) RT-PCR gel electrophoresis results of proband of F10 (BRCA2 c.793G>A carrier). No additional band different from the wild type is present. (F) Aligned sequencing electropherogram of cDNA and gDNA of the same region. Arrow points the variant c.793G>A. Neither an aberrant transcript, nor allelic imbalance is experienced. C1–3, wild type controls; F8, family 8; F10, family 10; gDNA, genomic DNA; L, 1 kb ladder (Promega); LOH, loss of heterozygosity; RT-PCR, reverse transcription polymerase chain reaction; tDNA, tumor DNA.
BRCA2 c.793G>A affected the last nucleotide of BRCA2 exon 9 and was prognosticated as potentially spliceogenic variant affecting canonical splice donor site by varSEAK program (score 4). Opposed to this prediction, we observed neither aberrant transcript nor allelic imbalance when tested cDNA of the variant carrier (Fig. 3E and F).
Discussion
This study based on 15 randomized controlled trials including 2,867 patients and aCorrect splicing regulation is indispensable for generating functional transcripts, so adequate evaluation of genetic variants’ role in aberrant splicing is of paramount clinical relevance. We analyzed five rare BRCA1/2 variants on cDNA-level, which were suggested a priori as potentially splice-altering changes according to various in silico splice predictions. Although RNA expression data arose from peripheral blood rather than tumor tissue samples of the carriers, data are authentical, since surveys give evidence that BRCA1 alternative splicing is similar in blood and breast tissue, a finding supporting the clinical relevance of blood-based in vitro splicing assays [25]. Besides the presence of aberrant splicing, the extent of that is also an issue in determining pathogenicity, since surveys argue that incomplete aberrant splicing may yield normal transcript in sufficient quantity for physiological function [26]. Since the analyzed RNAs were collected in Tempus Blood RNA tubes, we could not perform NMD-inhibition prior to cDNA analyses of the samples, but we were able to calculate its extent indirectly in most of the cases, where it was applicable. Utilizing tagging polymorphism is an acknowledged way of determining if the lower extent of alternative allele expression is the result of incomplete aberrant splicing or nonsense-mediated decay [27]. Additionally, as further steps, we investigated locus-specific LOH in breast cancer tumor tissues and also performed co-segration analysis of these variants with clinical phenotype.
Multiple lines of evidence were synthesized to prove pathogenicity using the standardized variant interpretation recommendations of the ACMG (Table 3). Of the variants studied, BRCA1 c.4484+4dupA and BRCA1 c.5407-10G>A had enough supportive evidence for reclassification from VUS into likely pathogenic (Tier 2) category. These main arguments are (1) multiple lines of computational evidence support a deleterious effect on the gene or gene product (PP3 evidence), (2) the variants were found in patients with disease phenotype (PP4), since the probands were young age at onset with personal disease phenotypes characteristic of BRCA1 mutation-carriers, in addition, BRCA1 c.4484+4dupA variant carrier families had several relatives having various tumors in the syndromic spectrum, (3) the variant alleles were absent in diverse variant databases such as Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org) or the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) (PM2) and (4) finally, our results fulfilled the PS3 category: ‘well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product are strong evidence for pathogenicity’ [4]. Our study provided transcript-level evidence for pathogenicity (pathogenic splice product, elicited unequivocally by the variant position), which was eligible for the assertion. Furthermore, CRISPR-based saturation genome editing surveys performed by Findlay et al. (2018) [23] also pointed out the possible functional relevance of BRCA1 c.5407-10G>A, with an intermediate functional score of −0.95. An additional layer of a posteriori evidence for pathogenicity was also provided by LOH test of the variant BRCA1 c.5407-10G>A, where the loss of the normal allele with R < 0.5 was demonstrated in the tumor tissues removed from both probands. Although the tumor sample of the proband carrying BRCA1 c.4484+4dupA did not show LOH, it is not a strong proof against pathogenicity, since the ACMG scoring system does not make use of the somatic results as independent evidence for clinical assertion [28]. Even the bona fide pathogenic BRCA1 mutations do not always accompanied by LOH. As much as 10% of the BRCA1 germline mutation-associated breast tumors did not show locus-specific LOH [29]. Co-segregation analysis, however, was achievable in a c.4484+4dupA variant carrier family, where the variant segregated with the phenotype in additional family members, corroborating its pathogenic nature.
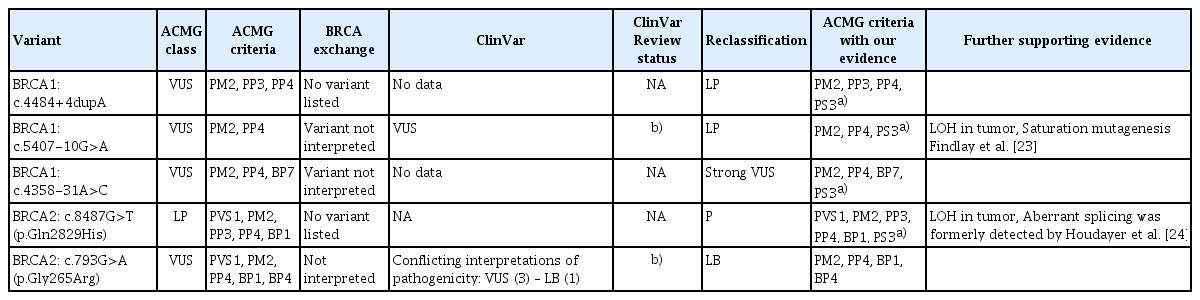
Variant classification
cDNA-level analysis of the variant BRCA1 c.4358-31A>C clearly showed the presence of aberrant splicing, which was also whole exon 14 skipping. This is most probably the consequence of the disturbance of the active branchpoint adenine in the splicing intermediate lariat formation. Nonetheless, indirect calculations based on the inferred extent of the NMD showed that the aberrant splicing is only partial, it totals up to only half of the transcripts of the variant-carrier allele. Functional surveys by de la Hoya et al. [26] revealed that BRCA-associated cancer risk is not markedly increased for individuals who carry a BRCA1 allele, which permits 20%–30% of tumor suppressor function. In contrast, Bonnet et al. (2008) [30] judged BRCA2 c.9501+3A-T variant with partial exon 25 skipping as a biologically significant mutation with reduced penetrance, although a significant portion of the variant-carrier allele produced normal transcript. Similarly, Zhang et al. (2009) [27] published that BRCA2 IVS4-12del5 is a mutation, though this variant causes only partial deletion of exon 5 as a result of inefficient aberrant splicing. In our case, the proband’s family (family 3) harbored strong characteristics of HBOC with five affected relatives, each fitting in the disease spectrum. Unfortunately, samples from other members of the family were not available for co-segregation analysis, similarly LOH test of the tumor was not feasible. In summary, this variant, although deserves attention, still requires further analysis to be equivocally asserted into the likely pathogenic category.
Transcript of the BRCA2 c.8487G>T variant allele showed complete exon 19 skipping in our study. The BRCA2 Δ19 is a minor naturally occurring alternative in-frame isoform but it is proved to be non-functional in complementation assays [31]. Spliceogenic capacity of this variant was formerly witnessed by Houdayer et al. [24] but they did not determine the amount of the aberrant splicing, which was assessed as 100% in our study. The other novelty provided by our experiments was the demonstration of LOH in several variant-carrier tumor samples, which is also corroborative for its pathogenic nature [28]. The variant is not registered in the dbSNP or ClinVar databases, but occurred relatively frequently in our familiar breast cancer cohort (4/3, 568). Phenotypes of the probands, as well as family tumor history, were characteristic for the pathology of BRCA1 carriers. ACMG scoring by VarSome ver. 2021 predicts this variant as likely pathogenic. Indeed, by the combined supportive evidence, we can reinforce this assertion.
As for the variant BRCA2 c.793G>A, as opposed to its spliceogenic prediction by varSEAK, our studies yielded neither aberrant transcript nor allelic imbalance at cDNA-level. The results provided sufficient evidence for this variant to alter the VUS ACMG verdict to likely benign.
Poor participant rate in family (cascade) screening is considered a limitation of the current study. While genetic testing was offered to all first-degree, asymptomatic and second-degree affected relatives during genetic counseling, in the 10 families only 14.8% (4/27) check-in rate was observed. Referred reasons from probands were elderly parents, living in different city or countryside and loose family bonds. Therefore, interpretation of co-segregation data has not represented strong relevance in our study.
As a summary, out of the five investigated variants, we were able to reclassify two VUS (BRCA1:c.4484+4dupA; BRCA1:c.5407-10G>A) into likely pathogenic class; one likely pathogenic variant (BRCA2:c.8487G>T, p.(Gln2829His)) into pathogenic category and one VUS (BRCA2:c.793G>A, p.(Gly265Arg)) into likely benign class.
With the spread of the high-throughput NGS in the routine molecular genetic diagnostics of hereditary cancer predisposition, there are emerging numbers of rare variants with unknown significance. The presence of VUS represents a significant challenge for the clinical geneticist, for the managing clinicians and for the patients as well. According to the current guidelines, VUS of the BRCA1/2 genes are reportable, however should not be used for medical decisions, which can result in considerable stress for the proband and the proband’s family. All of these emphasize the need for the molecular and clinical characterization of VUS. Both up- and down-classification harbor important clinical significance. Patients carrying re-classified pathogenic variants (previously known as VUS) in the future will not be dropped out from medical surveillance, preventive measures, treatment, and predictive family screening in relatives at risk. In the current study, we presented molecular and clinical evidence as a basis of reclassification and clinical evaluation of five BRCA1/2 variants that can be used in the interpretation of molecular genetic reports.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
The study was approved by the Institutional Ethical Board and the Research and Ethics Committee of the Hungarian Health Science Council (ETT-TUKEB 53720-7/2019/EÜIG) and performed in accordance with the principles of the Declaration of Helsinki. After genetic counseling, written informed consents were obtained from all patients.
Author Contributions
Conceived and designed the analysis: Bozsik A, Papp J, Grolmusz VK, Patócs A, Oláh E, Butz H.
Collected the data: Bozsik A, Papp J, Grolmusz VK, Patócs A, Oláh E, Butz H.
Contributed data or analysis tools: Bozsik A, Papp J, Grolmusz VK, Patócs A, Oláh E, Butz H.
Performed the analysis: Bozsik A.
Wrote the paper: Bozsik A, Papp J, Butz H.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgements
This work was supported by TKP2020-NKA-26 to A.P. and NKFI FK 135065 to H.B.