Arsenic Trioxide Induces Apoptosis in Human Colorectal Adenocarcinoma HT-29 Cells Through ROS
Article information
Abstract
Purpose
Treatment with arsenic trioxide (As2O3) results in a wide range of cellular effects that includes induction of apoptosis, inhibition of cell growth, promotion or inhibition of cellular differentiation, and inhibition of angiogenesis through a variety of mechanisms. The mechanisms of As2O3-induced cell death have been mainly studied in hematological cancers, and those mechanisms in solid cancers have yet to be clearly defined. In this study, the mechanisms by which As2O3 induces apoptosis in human colorectal adenocarcinoma HT-29 cells were investigated.
Materials and Methods
To examine the levels of apoptosis, HT-29 cells were treated with As2O3 and then we measured the percentage of Annexin V binding cells, the amount of ROS production and the mitochondrial membrane potential. Western blot analysis was performed to identify the activated caspases after As2O3 exposure, and we compared the possible target molecules of apoptosis. As2O3 treatment induced the loss of the mitochondrial membrane potential and an increase of ROS, as well as activation of caspase-3, -7, -9 and -10.
Results
As2O3 induced apoptosis via the production of reactive oxygen species and the loss of the mitochondrial membrane potential. As2O3 induced the activation of caspase-3, -7, -9 and -10. Furthermore, As2O3 treatment downregulates the Mcl-1 and Bcl-2 expressions, and the release of cytochrome c and an apoptosis-inducing factor (AIF). Pretreating the HT-29 cells with N-acetyl-L-cysteine, which is a thiol-containing antioxidant, inhibited the As2O3-induced apoptosis and caspase activation.
Conclusion
Taken together, these results suggest that the generation of reactive oxygen species (ROS) by As2O3 might play an important role in the regulation of As2O3-induced apoptosis. This cytotoxicity is mediated through a mitochondria-dependent apoptotic signal pathway in HT-29 cells.
INTRODUCTION
Arsenic trioxide (As2O3) has been used as a therapeutic agent for treating hypertension, bleeding gastric ulcers, heartburn, chronic rheumatism and cancer in the Orient countries (1). As2O3 has a long history of use for the treatment of leukemia, and it was recently approved for the treatment of relapsed or refractory acute promyelocytic leukemia (APL) by the US Food and Drug Administration (2). Treating APL patients with As2O3 is associated with the disappearance of the promyelocytic leukemia-retinoic acid receptor α (PML-RARα) fusion transcript; this is the gene product of the chromosomal translocation t (15;17), which is characteristic of APL (3). In addition, several in vitro studies revealed that As2O3 induces apoptosis in APL cells, multiple myeloma cells and some solid tumor cells such as prostate, ovarian carcinoma and neuroblastoma cells (4). Despite of the extensive investigations that have been conducted, the molecular mechanism of As2O3-induced apoptosis in solid tumors is not yet fully understood.
Programmed cell death or apoptosis is a physiological process that plays an important role in the maintenance of cellular homeostasis via the selective elimination of excessive cells (5). Apoptotic cells show the characteristic morphological changes that include cell shrinkage, chromatin condensation, nuclear fragmentation, membrane blebbing, caspase activation and the formation of membrane bound vesicles that are called apoptotic bodies (6). Aberrant regulation of apoptosis has been observed in many forms of human disease including cancers, AIDS and many neurodegenerative diseases (7). Therefore, understanding the mechanism of apoptosis has important implications for the prevention and treatment of many diseases.
Reactive oxygen species (ROS) play important roles in a variety of normal cellular biochemical functions and abnormal pathological processes. The biological functions of ROS and their potential roles in the development of cancers and disease progression have been investigated for decades (8~9). Evidence from recent studies suggests that cancer cells, as compared to normal cells, are under increased oxidative stress that is associated with the increased generation of ROS (10). Because ROS are chemically active and they can inflict severe cellular damage, the possibility that cancer cells are under increased intrinsic ROS stress may provide a unique opportunity to destroy malignant cells based on their vulnerability to further ROS insults (11).
Although the mechanisms of As2O3-induced cell death have mainly been studied in hematologic cancers, those studies have not clearly defined a role for As2O3 in solid tumors. In the present study, we have investigated As2O3's induction of apoptosis in human colorectal adenocarcinoma cells, and we have elucidated the regulatory mechanism of apoptosis via As2O3, by focusing on the generation of ROS.
MATERIALS AND METHODS
1) Materials
As2O3, MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazoliumbromide) and N-acetyl-L-cysteine (NAC) were obtained from Sigma (St. Louis, MO). Annexin V-FITC was purchased from Becton Dickson (San Diego, CA). The enhanced chemiluminescence (ECL) detection system was purchased from Amersham Biosciences (Buckinghamshire, UK). RPMI 1640 was purchased from Life Technologies, Inc. (Gaithersburg, MD), and fetal bovine serum was purchased from Hyclone (Logan, Utah). Antibodies against caspase-3, -7, -9 and -10 were obtained from Cell Signaling Technology (Beverly, MA) and the antibodies against Mcl-1, Bcl-2 and apoptosis-inducing factor (AIF) were from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against cytochrome c was purchased from Pharmigen (San Diego, CA), and antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from Chemicon (Temecula, CA).
2) Cell culture
HT-29 human colorectal adenocarcinoma cells were obtained from the American Type Culture Collection (Rockville, MD). The cells were maintained in RPMI 1640 supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin and 10% heat inactivated fetal bovine serum, and they were grown at 37℃ in 5% CO2 fully humidified air; they were subcultured twice weekly.
3) Cell viability assay
Cells (5×103 cells/well) were cultured in 96-well microplates and treated with As2O3 in the presence or absence of various concentrations of inhibitors. After incubating the plates for 48 h, 50 µl of MTT (2 mg/ml) was added and the plates were incubated for an additional 4 h under the same conditions. During the final 4 h of incubation, the yellow MTT tetrazolium salt is reduced to purple-blue formazan crystals in only the viable cells. Formazan crystals were dissolved by adding 250 µl DMSO and the absorbance, which is linearly related to the number of viable cells, was measured at 570 nm on a BioRad M450 microplate reader. The cell death rate was expressed as a percentage of untreated control cells.
4) Annexin V-FITC staining
To detect early apoptosis, the cells were stained with FITC-conjugated Annexin V. After treatment with or without As2O3, the cells were washed with cold PBS before being resuspended in a 100 µl cold binding buffer. Five microliters of Annexin V-FITC were added and the incubation was continued for 15 min at room temperature and in the dark. A further 400 µl of binding buffer were added to terminate the reaction; flow cytometry analysis was then performed immediately for at least 10,000 cells by using a fluorescence-activated cell-sorting (FACS) analysis system (Becton Dickinson, San Jose, CA).
5) Measurement of ROS production
Cells were treated with the indicated concentrations of As2O3 for 48 h. Cells were incubated in 10 µM 2',7'-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Molecular Probes, Eugene, OR) at 37℃ for 20 min. The cells were harvested by trypsinization and washed twice with cold PBS. The ROS were determined by FACS analysis.
6) Determination of mitochondrial membrane potentials
The mitochondrial membrane potentials were measured using JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide; Molecular Probes, Eugene, OR), which is based on staining live cells in a membrane potential-dependent fashion: green fluorescence (λem=527 nm) was the results of the JC-1 monomer and red fluorescence (λem=590 nm) was the result of the JC-1 aggregates (12). Briefly, the cells (3×105 cells/well) were treated with As2O3 for 48 h and then washed twice with PBS. The cell suspension was incubated with JC-1 (10 µg/ml) dye for 30 min at 37℃; the green (FL-1) and red (FL-2) fluorescence was then measured by FACS analysis.
7) Isolation of the cytosolic and mitochondrial fractions
The cells were washed once with cold PBS and then they were resuspended with 5 volumes of an isolation buffer (250 mM sucrose containing 20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol and 0.1 mM PMSF). After chilling on ice for at least 20 min, the cells were homogenized with 30~40 strokes of a Dounce homogenizer (70~80% cell breakage), and the homogenates were centrifuged at 750 g for 10 min at 4℃ to remove the unbroken cells and nuclei. The supernatants were harvested and centrifuged at 12,000 g for 30 min at 4℃ (the cytosolic fraction). The mitochondria-enriched pellets were resuspended in isolation buffer. The cytosolic and mitochondrial fractions were then subjected to electrophoresis and Western blot analysis.
8) SDS-PAGE and Western blot analysis
The cells were washed twice in cold PBS and lysed on ice with lysis solution (1% Triton X-100, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM PMSF and a protease inhibitor cocktail). The protein concentrations were determined using the BioRad protein assay according to the manufacturer's recommendations. The protein samples (30 µg of each) were separated on 10% SDS-polyacrylamide gel and electrotransferred to nitrocellulose membranes. The membranes were blocked for at least 30 minutes at room temperature in Tris-buffered saline plus 0.05% Tween-20 (TTBS) that contained 5% nonfat dry milk, and then the membranes were incubated with TTBS that contained a primary antibody. For immunoblotting, incubation was performed for 4 h at room temperature. After five washes within a 30 minute period, the membranes were incubated with a peroxidase-conjugated secondary antibody for 1 h. After five washes within a 30 minute period in TTBS, the membranes were developed using an ECL detection system and then they were exposed to X-ray films.
RESULTS
1) Effects of As2O3 on the apoptotic cell death of HT-29 cells
We examined the cytotoxic effect of As2O3 on human colorectal adenocarcinoma HT-29 cells. Treatment with As2O3 caused decreases in cell survival in a dose- and time-dependent manner (Fig. 1). To investigate whether the As2O3-induced cytotoxicity is mediated by apoptosis, we measured the Annexin V staining, ROS production, the mitochondrial membrane potential (Δψm) changes and the caspase cleavages in the cells that were treated with As2O3. Annexin V binds to the membrane phospholipid phosphatidylserine that is located within the plasma membrane of apoptotic cells. The percentage of cells stained with Annexin V was increased with the increasing concentrations of As2O3 (Fig. 2A). Since oxidative damage plays an important role in the anticancer effects of arsenic compounds (13), the intracellular generation of ROS in the HT-29 cells treated with As2O3 was investigated using an oxidation-sensitive fluorescent probe (H2DCF-DA), which is oxidized to 2',7'-dichlorofluorescein (DCF) in the presence of ROS. Treatment with As2O3 led to an increase of the DCF fluorescence in a dose-dependent manner (Fig. 2B). Since the production of ROS is usually preceded or accompanied with the loss of Δψm, we also examined whether As2O3 treatment induces the loss of Δψm in the HT-29 cells that had been treated with As2O3 by performing staining with JC-1. As expected, the As2O3 treatment induced the loss of the Δψm in a dose-dependent manner (Fig. 2C). The likely involvement of caspases in the As2O3-induced apoptosis of HT-29 cells was analyzed by performing Western blot analysis. The treatment of cells with As2O3 caused the activation of caspase-3, -7, -9 and -10 in a dose- and time-dependent manner, as indicated by a reduction in the intensity of the proenzymes (Fig. 2D). In addition, degradation of PARP, which a well-known substrate of caspase-3, increased in the cells treated with As2O3. However, the expression level of caspase-8 between the As2O3-treated and As2O3-untreated cells was not altered (data not shown). These results suggest that As2O3 might induce apoptotic cell death in HT-29 cells.

Effects of As2O3 on cell survival in human colorectal adenocarcinoma HT-29 cells. The cells were treated with increasing concentrations of As2O3. Cell survival was measured by the MTT assay. The values are the mean±SD of three independent experiments.

Induction of apoptosis by As2O3-mediated cytotoxicity. The cells were treated with increasing concentrations of As2O3 for 48 h. The cells were incubated with Annexin V (A), H2DCF-DA (B), or JC-1 (C), and then they were analyzed by flow cytometry. The cell lysates were further analyzed by Western blotting with using anti-caspase and anti-PARP antibodies (D). The GAPDH level was used to demonstrate equal loading of proteins on each lane.
2) Analysis of mitochondrial participation in As2O3-induced apoptosis
A key event in the apoptotic process is the opening of the mitochondrial permeability transition pores, and this is an event that's known to be regulated by the Bcl-2 family of proteins. The outcome of this opening is the release of several proteins such as procaspase-2, -3 and -9, cytochrome c and AIF from the mitochondrial intermembrane space (14). The expression level of Mcl-1 protein was significantly decreased in the As2O3-treated cells and the Bcl-2 protein level was slightly decreased (Fig. 3A). The release of cytochrome c from the mitochondria was also increased in the As2O3-treated cells compared to the untreated cells. The cells treated with As2O3 showed an increased amount of released AIF from the mitochondria into the cytosol (Fig. 3B). These results suggest that As2O3 might induce cellular apoptosis through a mitochondria-dependent apoptotic pathway.

Effects of As2O3 on the levels of Mcl-1, Bcl-2, cytochrome c and AIF. The cells were treated with increasing concentrations of As2O3 for 48 h. The cell lysates were analyzed by Western blotting using anti-Mcl-1, anti-Bcl-2, anti-cytochrome c and anti-AIF antibodies. The GAPDH level was used to demonstrate equal loading of proteins on each lane.
3) Effect of antioxidant on the apoptotic activity of As2O3
To determine a link between the elevation of the intracellular ROS level and the apoptotic cell death in the As2O3 treated cells, HT-29 cells were incubated with NAC prior to their treatment with As2O3. NAC pretreatment resulted in protection against the As2O3-induced cytotoxicity (Fig. 4). The As2O3-induced increases of Annexin V staining, the intracellular ROS levels and the Δψm were reduced by the NAC pretreatment (Fig. 5A and 5B). Furthermore, NAC inhibited the As2O3-induced caspase activation and PARP degradation (Fig. 5C). Yet the addition of NAC did not affect the As2O3-induced down-regulation of Mcl-1 and Bcl-2 (Fig. 5D). These results suggest that an increase in the intracellular ROS level by As2O3 treatment is required for the As2O3-induced apoptosis that is accompanied by the loss of Δψm, caspase activation and PARP degradation.
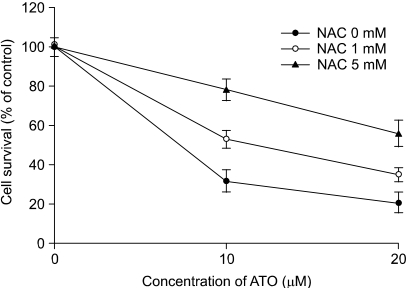
The effects of NAC on the As2O3-mediated cytotoxicity. The cells were treated with As2O3 in the presence or absence of NAC. After 48 h, cell survival was determined by the MTT assay. The values are the mean±SD of three independent experiments.

The effects of NAC on As2O3-mediated apoptosis. The cells were treated with 5 mM NAC for 30 min and then they were treated with 20 µM As2O3 for an additional 48 h. The cells were incubated with Annexin V (A), H2DCF-DA (B), or JC-1 (C) and next analyzed by flow cytometry. The cell lysates were analyzed by Western blotting with anti-caspase and anti-PARP antibodies (D). The GAPDH level was used to demonstrate equal loading of proteins on each lane.
DISCUSSION
The aim of the present study was to elucidate the molecular mechanisms of cellular death that are triggered by As2O3 treatment with using human colorectal adenocarcinoma HT-29 cells. Our experiments indicated that ROS have an important role in the As2O3-induced caspase-dependent cell death of the HT-29 cells.
The caspase cascade is the most-studied and best-understood signaling pathway in apoptosis. These proteases are produced as forms of pro-enzymes, and they can be activated by proteolysis in response to a diverse set of apoptotic stimuli (15). One means of activating the caspase cascade is termed the "death-receptor pathway" and this is initiated at the cell surface by the binding of apoptotic molecules to their receptors (16). Caspase-8 and caspase-10 are the key initiator caspases in the "death-receptor pathway". The mitochondria are another site for apoptotic stimuli to initiate an intracellular signaling process that mediates caspase activation. In this case, caspase-9 functions as an initiator caspase that activates downstream effecter caspases such as caspase-3 (17). We found that cells treated with As2O3 show activation of caspase-3, -7, -9 and -10. Degradation of PARP was also observed in the As2O3-treated cells. Thus, these data suggest that the As2O3-induced apoptosis of HT-29 cells might be mediated through a mitochondria-dependent pathway.
The mitochondria have been shown to play a pivotal role in the apoptotic process since both the intrinsic and the extrinsic pathways can converge at the mitochondrial level and so trigger changes in the mitochondrial membrane permeability (18,19). Mutations in such apoptosis-regulatory genes as p53, PTEN and Bcl-2 and its homologues are involved in the pathogenesis of most human cancers (20). Similar mutations may also be involved in the development of chemoresistance. Bcl-2 has been shown to be protective against diverse cytotoxic insults, i.e., UV-irradiation and cytokine drugs, while the pro-apoptotic family members may also act as tumor suppressors (21). Mcl-1 also has sequence homology to Bcl-2 and it further resembles Bcl-2 since its expression promotes cell viability (22). It has been reported that AIF mediates mitochondria-related cell death through a caspase-independent pathway wherein the mitochondrial AIF translocates to the cell nucleus in response to death stimuli, and there it initiates nuclear condensation. Once the nucleus condenses, large-scale chromatin fragmentation ensues followed by death of the cell (4). In this study, As2O3 induced the release of cytochrome c and AIF, so it's possible that As2O3 induces mitochondria-mediated apoptosis by both the caspase-dependent and caspase-independent pathways.
Accumulation of intracellular ROS leads to the disruption of the Δψm, the release of cytochrome c with the subsequent activation of the caspase cascade, and ultimately to programmed cell death via apoptosis (23,24). Arsenic exposure leads to increases in the ROS levels by a variety of redox enzymes such as NADPH oxidase (25). In the present study, we showed that increased levels of ROS are essential for the loss of the Δψm and the caspase activation that's triggered by As2O3. The significant inhibition of As2O3-induced caspase activation and cell death after NAC treatment suggests that increased ROS levels are critical for As2O3-mediated apoptosis in HT-29 cells.
CONCLUSIONS
The data presented in this study suggest that the generation of ROS might be a critical factor for As2O3-induced apoptosis in human colorectal adenocarcinoma HT-29 cells, and that the As2O3-induced apoptosis could be induced by a mitochondria-mediated apoptotic pathway.
Notes
This study was supported by the Korea Science and Engineering Foundation (KOSEF) through the Aging-associated Vascular Disease Research Center at Yeungnam University (R13-2005-005-01003-0 (2005).