Enhanced Efficacy of Combined Therapy with Checkpoint Kinase 1 Inhibitor and Rucaparib via Regulation of Rad51 Expression in BRCA Wild-Type Epithelial Ovarian Cancer Cells
Article information
Abstract
Purpose
This study aimed to evaluate anticancer effects of combination treatment with poly(ADP-ribose) polymerase (PARP) and checkpoint kinase 1 (Chk1) inhibitors in BRCA wild-type ovarian cancer. PARP inhibitors can function as DNA-damaging agents in BRCA wild-type cancer, even if clinical activity is limited. Most epithelial ovarian cancers are characterized by a TP53 mutation causing dysfunction at the G1/S checkpoint, which makes tumor cells highly dependent on Chk1-mediated G/M phase cell-cycle arrest for DNA repair.
Materials and Methods
We investigated the anticancer effects of combination treatment with prexasertib (LY2606368), a selective ATP competitive small molecule inhibitor of Chk1 and Chk2, and rucaparib, a PARP inhibitor, in BRCA wild-type ovarian cancer cell lines (OVCAR3 and SKOV3).
Results
We found that combined treatment significantly decreased cell viability in all cell lines and induced greater DNA damage and apoptosis than in the control and/or using monotherapies. Moreover, we found that prexasertib significantly inhibited homologous recombination–mediated DNA repair and thus showed a marked anticancer effect in combination treatment with rucaparib. The anticancer mechanism of prexasertib and rucaparib was considered to be caused by an impaired G2/M checkpoint due to prexasertib treatment, which forced mitotic catastrophe in the presence of rucaparib.
Conclusion
Our results suggest a novel effective therapeutic strategy for BRCA wild-type epithelial ovarian cancer using a combination of Chk1 and PARP inhibitors.
Introduction
Epithelial ovarian cancer is the most lethal gynecologic malignancy, leading to more than 140,000 deaths per year worldwide [1]. In South Korea, estimated new cases of ovarian cancer are 2,941 and estimated deaths from ovarian cancer are 1,309 in 2020 [2]. More than 80% of patients experience recurrences and more than half show chemo-resistance. Therefore, it is critical to understand and overcome the mechanism(s) involved in chemo-resistance in order to develop better therapeutic strategies.
The DNA damage repair (DDR) pathway, an extensive network of pathways that detects and signals DNA damage for subsequent processing, is known as a key process in human cancer development [3]. Ataxia telengiectasia and Rad3-related (ATR) and its downstream kinase, checkpoint kinase I (Chk1), are major components of the DDR pathway [4]. Most high-grade serous ovarian cancers (HGSC) show a mutation in TP53, which results in the loss of G1 checkpoint control, and thus significantly rely on S and G2 checkpoints for survival [5,6]. Therefore, targeting S and G2 checkpoints by inhibition of the ATR/Chk1 pathway in a tumor with a TP53 mutation will prevent DNA damage-induced G2 checkpoint arrest, leading to mitotic catastrophe and tumor cell death [7].
Poly(ADP-ribose) polymerase (PARP) inhibition, which leads to the failure of double strand break (DSB) repair in BRCA1 and BRCA2 defective cells, is another strategy targeting the DDR pathway. BRCA1 and BRCA2 are essential in homolgous recombinant repair of DSBs. Approximately 15% of patients with HGSC harbor deleterious germ-line mutations in BRCA1 and BRCA2 [8]. By inhibiting PARP in ovarian cancer with BRCA mutations, a failure of DSB repair, promotion of genomic instability, apoptosis, and cancer cell death will occur [9]. In a clinical setting, PARP inhibitor monotherapy have shown modest activity even in BRCA wild-type HGSC [10,11]. The limited activity of a PARP inhibitor against BRCA wild-type HGSC is partly caused by the overexpression of Rad51, which is associated with chemo-resistance [12].
Chk1 is known to play a critical role in homologous recombinant DNA repair. Chk1 facilitates the BRCA2-Rad51 interaction by phosphorylation of the BRCA2 C-terminal domain and Rad51 at T 309. This is an important step that allows the transnuclear localization of HR repair proteins in response to DSBs [13,14].
We hypothesized that inhibiting Chk1 would sensitize BRCA wild-type HGSCs to a PARP inhibitor by preventing the formation of Rad51 foci. In this study, we evaluated the preclinical efficacy of prexasertib (LY2606368), a selective ATP competitive small molecule inhibitor of Chk1 and Chk2, in combination with rucaparib, a PARP inhibitor, at clinically attainable concentrations in BRCA wild-type ovarian cancer.
Materials and Methods
1. Cell culture and drugs
SKOV-3 and OVCAR-3 (BRCA wild type) cell lines were purchased from the American Type Culture Collection (Rockville, MD). SKOV-3 cell lines were cultured in McCoy’s 5A medium (Welgene, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S; Invitrogen, Carlsbad, CA) in a humidified chamber with 5% CO2 at 37°C. OVCAR-3 cells were cultured in RPMI 1640 medium (Roswell Park Memorial Institute, Buffalo, NY) supplemented with 10% FBS. Prexasertib and rucaparib were purchased from Selleckchem (Houston, TX) and dissolved in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO). Final concentrations in culture medium never exceeded 0.2%.
2. Cell viability
Cell viability was measured by PrestoBlue cell viability reagent (Invitrogen). SKOV3 (1×105 cells/well) and OVCAR-3 cells (1×105 cells/well) were plated in 96-well plates in McCoy’s 5A and RPMI completed media respectively. Each cell lines were treated with prexasertib, rucaparib, and both combination at 0, 1, 5, 10, 50, and 100 μM for 72 hours. The treated cells were incubated with 10% PrestoBlue reagent for 30 minutes in room temperature. The absorbance at 540 nm was measured by enzyme-linked immunosorbent assay microplate reader (Molecular Devices, San Jose, CA). All data are expressed as a percentage of control.
3. Cell proliferation
Cell proliferation was examined using a Cell Titer-Glo assay kit (Promega, Madison, WI). Cells were seeded in 96-well microplates in McCoy’s 5A complete medium (containing 10% FBS and 1% P/S) and treated with prexasertib, rucaparib or a combination at 0, 1, 5, 10, 50, and 100 μM for 72 hours. A volume of Cell Titer-Glo reagent equal to the volume of cell culture medium was added to cells in each well. Cells were incubated at room temperature for 10 minutes and a luminescent signal was recorded by a luminescence microplate reader (Berthold Technologies, Bad Wildbad, Germany).
4. Rad51 siRNA transfection
Human Rad51 siRNA was synthesized by Genolution (Seoul, Korea), and siRNA for control was purchased from Santa Cruz Biotechnology (Dallas, TX). The Rad51 targeting-sequence was 5′-AAGCUGAAGCUAUGUUCGCCAUU-3′. The transfection was performed using Opti-MEM media and Lipofectamine RNAi MAX (Invitrogen) according to the manufacturer’s method. Transfected cells were cultured in 96-well plates or 100-mm culture dishes. At 48 hours after transfection, the cells were treated with rucaparib 50 μM for 48 hours, and then cell viability and caspase-3 activity were determined or an immunoblot was undertaken.
5. Apoptosis by annexin V–FITC by flow cytometry
For apoptotic cell death analysis, an annexin V–FITC assay was carried out using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the protocol provided. The cells, at a concentration of 1×106 cells/well, were seeded in a 6-well plate and treated with 0, 50, and 100 μM concentrations of prexasertib, rucaparib or a combination of these for 48 hours. After treatment, supernatants and cells were harvested and centrifuged at 1,200 rpm for 7 minutes. The cell pellet was resuspended in 100 μL of 1× binding buffer, and 5 μL of FITC Annexin V and propidium iodide (PI) were added. Cells were incubated for 15 minutes at room temperature (25°C) in the dark. After incubation, 1× Binding Buffer was added to each sample, and cells were analyzed by flow cytometry within 1 hour. Flow cytometric analysis was carried out using a FACS Calibur (BD Biosciences) flow cytometer, by analyzing at least 10,000 cells per sample. Results are presented as a percentage of the total gated number of cells.
6. Cell-cycle arrest by flow cytometry
For cell-cycle analysis, treated cell samples were washed with ice-cold phosphate-buffered saline (PBS) and fixed with 70% ethanol at −20°C overnight. Then, samples were washed with PBS and resuspended with 0.5 mL of FxCycle PI/RNase Staining Solution (Invitrogen) containing 50 μg/mL PI with 100 μg/mL RNase A, and incubated for 30 minutes at room temperature while protected from light. Samples were analyzed by FACS Calibur (BD Biosciences) flow cytometer.
7. Caspase 3/7 activity
SKOV-3 or OVCAR-3 cells (1×105/well) in a white-walled 96-well plate were cultured for 24 hours in McCoy’s 5A or RPMI 1640 complete media, and treated with prexasertib, rucaparib or a combination for 48 hours. The treated cells were incubated with 100 μL of Caspase-Glo 3/7 reagent at room temperature for 30 minutes. The luminescence of each sample was measured in by a luminometer (Molecular Devices). All data are expressed as a fold induction of control.
8. Western blot analysis
Treated cells were lysed by ice-cold cell lysis buffer (Intron Biotechnology, Seongnam, Korea), and protein concentrations were determined with a BCA assay kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Equal amounts of protein (20–30 μg/lane) was separated on a 12% acrylamide gel by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane, and blocked with 5% non-fat milk. Each membrane was incubated with anti-Chk1, anti–phospho Chk1 (Ser345), anti–phospho Chk1 (Ser296), anti-PARP, anti–cyclin B1 (Cell Signaling Technology, Danvers, MA); anti-Rad51, anti–γ-H2AX (Ser139), anti–phospho histone H3 (Ser10), anti–cleaved caspase-3, anti–cleaved PARP (Abcam, Cambridge, UK); and alpha-tubulin (Sigma-Aldrich) antibodies. Each membrane was then incubated with horse radish peroxidase (HRP)–conjugated secondary anti-mouse or rabbit IgG antibody, and protein bands visualized using Immobilin Forte Western HRP Substrate (Millipore, Burlington, MA).
9. Statistical analysis
All procedures were performed using at least 16 samples and repeated in three independent experiments. All data are represented by the mean value±standard deviation. Since, Komogorov-Smmirnov test revealed that all variables showed normal distribution, student’s t test was used to determine the p-value between the two different groups. A p-value of less than 0.05 was considered significant.
Results
1. Chk1 inhibition induces apoptotic cell death compared to PARP inhibition in BRCA wild-type ovarian cancer cell lines
First, to validate the induction of cell death by Chk1 or PARP inhibition in ovarian cancer cell lines, we evaluated cell viability and proliferation in the presence of prexasertib and rucaparib using PrestoBlue and Cell Titer-Glo assays. The cell viability and proliferation of SKOV-3 and OVCAR-3 cells were reduced when cells were treated with prexasertib compared to rucaparib (Fig. 1A and B). Caspase-3 luciferase activity involved in the apoptotic signaling pathway increased in a dose-dependent manner in prexasertib-treated SKOV-3 and OVCAR-3 cells, but not in rucaparib-treated cells (Fig. 1C). Immunoblot analysis revealed that cells increased the expression of the pro-apoptotic proteins, cleaved caspase-3, and cleaved PARP, under prexasertib treatment (Fig. 1D). These results suggest that Chk1 inhibition induced apoptotic cell death in ovarian cancer cells.
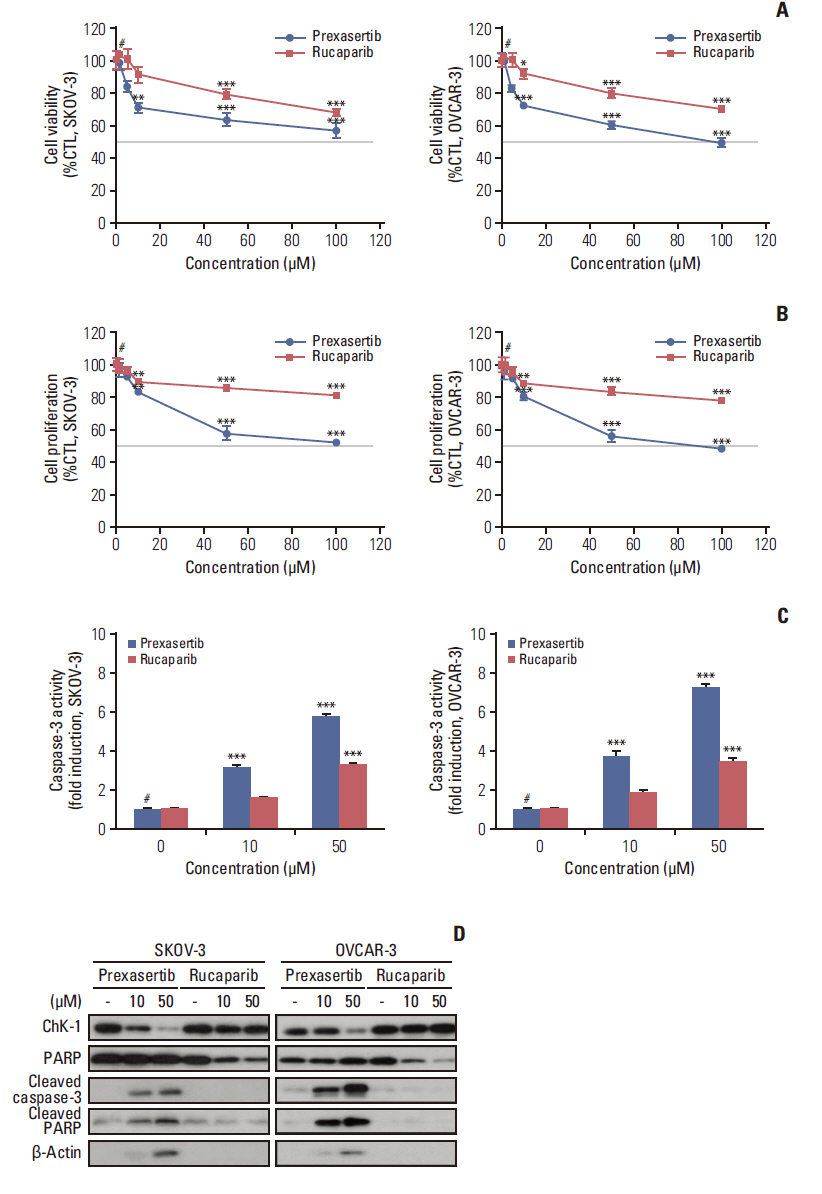
Prexasertib or rucaparib reduce cell viability and proliferation and induce apoptotic cell death in BRCA wild-type ovarian cancer cell lines. The cells were treated with either prexasertib (0–100 μM) or rucaparib (0–100 μM) for 72 hours after cells were seeded. The cell viability (A) and proliferation (B) of prexasertib and rucaparib was determined by PrestoBlue or Cell Titer-Glo assay in SKOV-3 and OVCAR-3 cells. (C) The activity of caspase-3 was measured by luciferase assay using Caspase-Glo 3/7 reagent. Cell viability, proliferation, and caspase-3 activity were calculated relative to 0.01% dimethylsulfoxide-treated control cells. (D) Representative images of immunoblotting data for protein levels of checkpoint kinase 1 (Chk1), poly(ADP-ribose) polymerase (PARP), cleaved caspase-3, and cleaved PARP proteins after prexasertib or rucaparib treatment. Alpha-tubulin was used as a loading control. Blue and red colors indicate prexasertib and rucaparib, respectively. Values are expressed as the mean±standard deviation. *p < 0.05 compared to 0 μM, **p < 0.01 compared to 0 μM, ***p < 0.001 compared to 0 μM, #Control group (CTL).
2. Chk1 inhibition combined with PARP inhibition promotes apoptotic cell death in BRCA wild-type ovarian cancer cell lines
We next assessed the combined effect of Chk1 and PARP inhibition in ovarian cancer. Cell viability curves for combination treatments, ranging from 0–100 μM for prexasertib, and 0, 10, and 50 μM for rucaparib, were determined from PrestoBlue assays (Fig. 2A). We then selected two doses of prexasertib/rucaparib (10, 50 μM/10 μM and 10, 50 μM/50 μM) to test for a combination effect. Apoptosis and caspase-3 activity analysis demonstrated that combination therapy increased apoptotic cell death compared to prexasertib or rucaparib monotherapy in both SKOV-3 and OVCAR-3 cells (Fig. 2B and C). Western blot analysis showed that combination therapy promoted the expression of cleaved caspase-3 and cleaved PARP, compared to prexasertib or rucaparib monotherapy, in a dose-dependent manner in ovarian cancer cells (Fig. 2D). This suggests that combined Chk1 and PARP inhibition, compared to the respective monotherapies, induced apoptotic cell death in ovarian cancer cells.

Combination treatment promotes suppression of cell viability and induction of apoptotic cell death in ovarian cancer cells. SKOV-3 and OVCAR-3 cells were treated with either prexasertib (0–100 μM) or rucaparib (0, 10, and 50 μM) for 72 hours after cells were seeded. (A) Cell viability was by PrestoBlue in SKOV-3 and OVCAR-3 cells. (B) Apoptotic cell analysis was measured using an Annexin V assay by fluorescence activatdetermineded cell sorting. (C) Caspase-3 activity was measured by luciferase assay using Caspase-Glo 3/7 reagent. (D) Representative images of immunoblotting data for levels of checkpoint kinase 1 (Chk1), poly(ADP-ribose) polymerase (PARP), cleaved caspase-3, and cleaved PARP proteins in combination treatment conditions. Alpha-tubulin was used as a loading control. *p < 0.05 compared to 0 μM, **p < 0.01 compared to 0 μM, ***p < 0.001 compared to 0 μM, #Control group. Values are expressed as the mean±standard deviation.
3. Chk1 inhibition could force mitotic entry of G2M-phase cells induced by PARP inhibitor in BRCA wild-type ovarian cancer cell lines
Cell-cycle analysis revealed that prexasertib could force mitotic entry of SubG1 phase cells induced by rucaparib in BRCA wild-type ovarian cancer cells (Fig. 3A). SubG1 phase cells, which suggested the presence of an apoptotic population, were significantly increased after combination therapy in SKOV-3 and OVCAR-3 cell lines. Western blot analyses showed that the expression level of the mitotic marker, p-H3, increased after combination therapy, which suggested that the blockade was in fact in M phase (Fig. 3B). Moreover, prexasertib-treated cells showed a substantial reduction in the level of endogenous Chk1, whereas phosphorylation of Chk1 (Ser345) and expression of γ-H2AX was significantly increased, indicating the persistence of DSBs in the treatment group (Fig. 3B). Therefore, our results suggest that combination therapy forced mitotic entry of G2M phase cells with unrepaired DNA damage, which may have led to a synergistic anticancer effect compared to monotherapy of each drug.
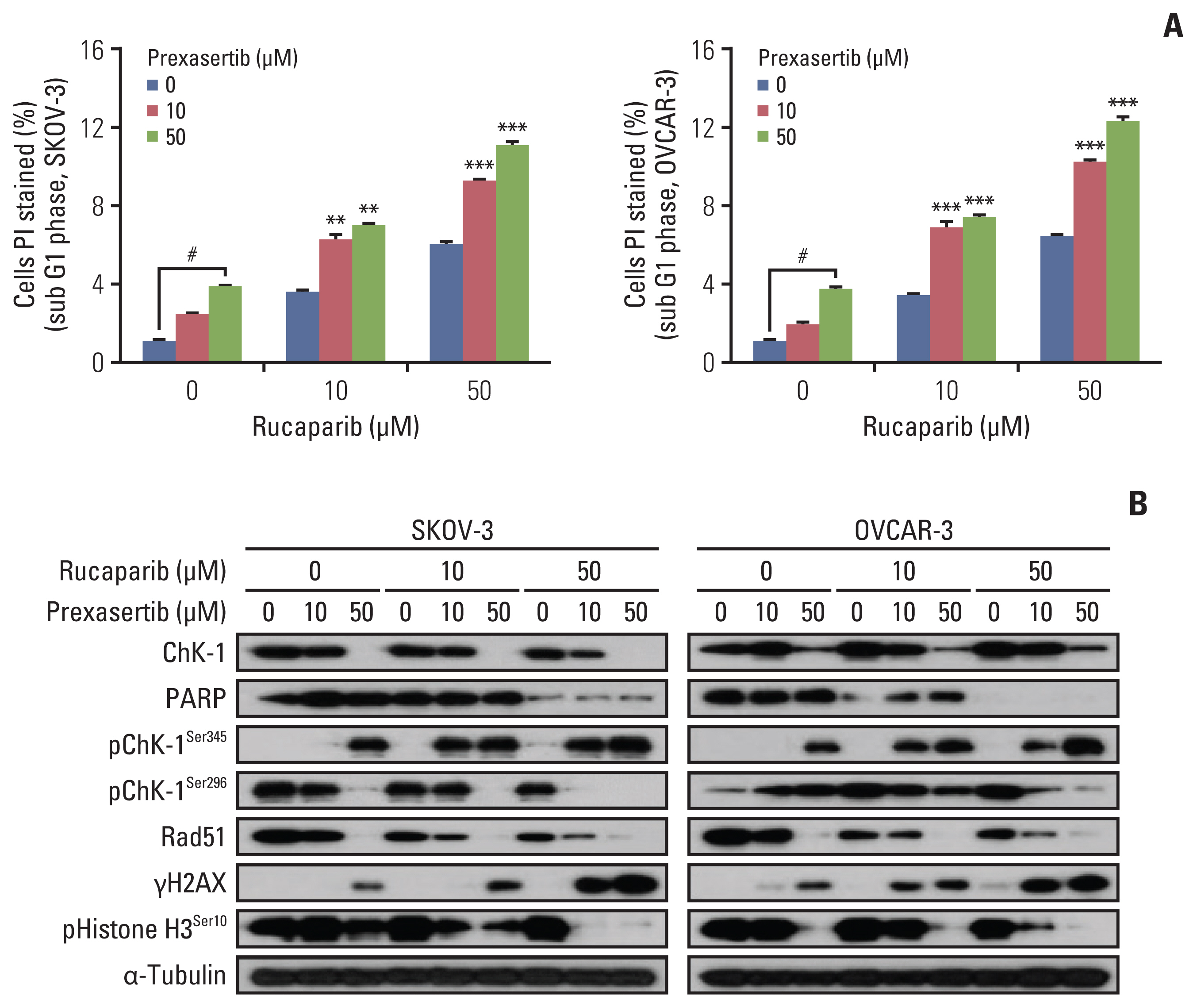
Combination treatment increases mitotic entry of sub G1 phase cells and activates the DNA damage signaling pathway. (A) Cell-cycle analysis was determine using FxCycle propidium iodide (PI)/RNase staining solution by fluorescence activated cell sorting in both SKOV-3 and OVCAR-3 cell lines. (B) Representative images of immunoblotting data for levels of phospho-Ser345 checkpoint kinase 1 (Chk1), phospho-Ser296 Chk1, Rad51, γH2AX, and phosphor-Ser10 Histone H3 proteins in combination treatment conditions. Alpha-tubulin was used as a loading control. Values are expressed as the mean±standard deviation. **p < 0.01 compared to 0 μM, ***p < 0.001 compared to 0 μM, #Control group.
4. Chk1 inhibition prevents nuclear Rad51 foci formation in response to rucaparib treatment in BRCA wild-type ovarian cancer cell lines
Gene expression data through analysis of The Cancer Genome Atlas was evaluated; this revealed Rad51 was a poor prognostic marker for patients with breast cancer. Additionally, Rad51 expression levels were much higher in African–American and Asian patients with breast cancer compared to Caucasians, suggesting Rad51 was a biomarker for racial disparities in this disease. Also, our immunoblotting data revealed that Rad51 expression was increased and regulated depending on Chk1 expression in ovarian cancer cells (Fig. 3B). Therefore, to evaluate the anticancer effect in response to inhibition of Rad51, we incorporated siRad51 in both cell lines and treated these with rucaparib. Cell viability reduced Rad51 knockdown and rucaparib treatment (Fig. 4A); caspase-3 activity was induced under the same conditions in both SKOV-3 and OVCAR-3 cells (Fig. 4B). Western blot analyses of BRCA wild-type ovarian cancer cell lines treated with siRad51 and rucaparib showed a similar pattern to those of cells treated with prexasertib and rucaparib, indicating that Chk1 inhibition by prexasertib prevents nuclear Rad51 foci formation in response to rucaparib treatment (Fig. 4C). These results demonstrated that synergistic cytotoxicity in combination treatment with prexasertib and rucaparib is caused by a reduced Rad51 response. The induction of nuclear Rad51 foci by rucaparib might be abrogated when Chk1 is inhibited by prexasertib in BRCA wild-type ovarian cancer cell lines.

Regulation of Rad51 expression affects anticancer effect by poly(ADP-ribose) polymerase (PARP) inhibition. (A, B) Knockdown of Rad51 expression was performed by Lipofectamine in ovarian cancer cells. After inhibition of Rad51, SKOV-3, and OVCAR-3 cells were treated with 50 μM rucaparib for 48 hours. Cell viability and caspase-3 activity were measured by PrestoBlue and Cell Titer-Glo assays, respectively. (C) Representative images of immunoblotting data for protein levels of Rad51, checkpoint kinase 1 (Chk1), PARP, γH2AX, cleaved caspase-3, and cleaved PARP under specific conditions. Alpha-tubulin was used as a loading control. Values are expressed as the mean±standard deviation. **p < 0.01 compared to 0 μM, ***p < 0.001 compared to 0 μM, #Control group.
Discussion
Currently, PARP inhibitors such as olaparib, rucaparib, and niraparib, have been approved by the U.S. Food and Drug Administration and shown to have clinical potential in treating ovarian cancer. However, olaparib monotherapy achieved only a 30% response rate for the treatment of BRCA wild-type ovarian cancer [15].
Individual PARP inhibitors have different binding affinities for PARP1, PARP2, and PARP3 [16]. Thus, on-target effects might be different according to the types of PARP inhibitors [16]. Rucaparib inhibits PARP1, PARP2, and PARP-3, whereas olaparib and niraparib inhibits only PARP1 and PARP2 [16]. In addition, PARP3 has been reported to activate the enzymatic activity of PARP1 in the absence of DNA. Therefore, the additional inhibition of PARP3 might potentiate the effects of rucaparib compared with olaparib or niraparib [16,17].
A part 1 of the ARIEL2 trial revealed that rucaparib monotherapy was efficacious in women with relapsed, platinum-sensitive, BRCA mutated HGSC cell lines, as well as in those with BRCA wild-type carcinomas with high genomic loss of heterozygosity, a potential marker of homologous recombination deficiency and PARP inhibitor activity [18,19]. In our study, we applied rucaparib as a PARP inhibitor, expecting to have compatible potency with olaparib or niraparib, as well as comparable interaction with Chk1 inhibitor in BRCA wild-type ovarian cancer.
To achieve a higher complete response for the treatment of BRCA wild-type ovarian cancer, a combination strategy with PARP inhibitors and other cytotoxic agents has been attempted. From this viewpoint, the ATR/Chk1 axis can be an attractive target. Several studies reported that inhibition of the ATR/Chk1 axis caused replication catastrophe, DNA damage, and cell death [20]. Moreover, several studies have reported that Chk1 can overcome the chemo-resistance of PARP inhibitors, and synergizes cytotoxic effects in cancer cells, including in ovarian, mammary, and gastric cancer cell lines [4,21–23].
In this study, we demonstrated that a combination of prexasertib with the PARP inhibitors, rucaparib, showed synergistic cytotoxicity against BRCA wild-type HGSC cell lines. First, we found that monotherapy of each drug, especially the Chk1 inhibitor, significantly suppressed cell proliferation. Nonetheless, combination therapy showed a synergistic effect in the suppression of cell proliferation and cytotoxicity. Our results are similar to those of several prior studies. A study exists that evaluates the in vitro toxicity of the PARP inhibitors, olaparib, in combination with prexasertib for the treatment of BRCA mutant and BRCA wild-type high-grade serous ovarian cancer (OVCAR3, OV90, PEO1, and PEO4) cell lines [23]. The authors suggested that combination treatment synergistically decreased cell viability in all cell lines, and induced greater DNA damage and apoptosis than monotherapy of each drug (p < 0.05 for all) [23]. In addition, they demonstrated that treatment with olaparib in BRCA wild-type HGSC cell lines caused the formation of Rad51 foci, whereas combination treatment with prexasertib inhibited the transnuclear localization of Rad51 [23]. Rad51 is a key protein in homologous recombination [23]. Therefore, they suggested that prexasertib increased the cytotoxicity of PARP inhibitor by preventing Rad51 foci formation in BRCA wild-type HGSC cell lines [23]. In our study, we used rucaparib which are expected to have compatible potency with other PARP inhibitors, such as olaparib and niraparib. Similar to prior study using olaparib, combination of Chk1 inhibitor and rucaparib also showed synergistic anticancer effect in BRCA wild-type ovarian cancer cell lines [23]. The authors reported that olaparib treatment induced nuclear Rad foci formation in BRCA wild-type HGSC cell lines, while prexasertib had no impact on nuclear Rad51 foci formation [23]. They hypothesized that the induction of nuclear Rad51 foci by olaparib was completely abrogated when Chk1 is inhibited by prexasertib in all BRCA wild-type HGSC cell lines [23]. On the contrary, in our study, western blotting showed that Rad51 expression decreased under prexasertib monotherapy and a combination of prexasertib and rucaparib, whereas it increased under rucaparib monotherapy, which might be comparable to prior study using olaparib. Furthermore, we proved that reduced levels of Rad51 expression by siRNA increases sensitivity to rucaparib in BRCA wild-type ovarian cancer cell lines. Our study confirms that Chk1 potentiates sensitivity to PARP inhibitor of BRCA wild-type ovarian cancer cells by suppression of Rad51. In addition, our data support that the interaction mechanism of Chk1 inhibitor and PARP inhibitor was similar, regardless of types of PARP inhibitors.
Another study evaluated the cytotoxic effect of a combination of PARP inhibitor and ATR inhibitor/Chk1 inhibitor in BRCA mutant ovarian cancer cell lines [4]. It was suggested that a combination of PARP inhibitor with ATR/CHK1 inhibitor is more effective than PARP inhibitor monotherapy in BRCA mutant ovarian cancer cell lines due to the increased reliance on ATR/CHK1 for genome stabilization under PARP inhibitor treatment [4].
Another study reported that a Chk1 inhibitor potentiated the cytotoxic effect of PARP inhibitor in gastric cancer cell lines [22]. The authors showed that a Chk1 inhibitor inhibits homologous recombination–mediated DNA repair, and thus had a marked synergistic anticancer effect in combination with PARP inhibitor in both in vitro studies and in vivo experiments, using a gastric cancer patient-derived xenograft model [22]. They suggested that synergy between the Chk1 inhibitor, LY2606368, and PARP inhibitor might be caused by an impaired G2M checkpoint due to LY2606368 treatment, which forced mitotic entry and cell death in the presence of a Chk1 inhibitor [22].
In conclusion, we demonstrate that a Chk1 inhibitor suppresses Rad51, which affects a decrease in homologous recombinant repair. Moreover, we found that Chk1 inhibitor and PARP inhibitor combination therapy forced mitotic catastrophe and cell death in p53-mutated ovarian cancer cell lines, which are highly dependent on G2/M phase cell-cycle arrest.
Importantly, we found that suppression of Rad51 sensitized cells to the anticancer effect of the PARP inhibitor, which might be applied to the treatment of various human cancer cells. This provides a potentially new therapeutic strategy for the treatment of BRCA wild-type HGSC, which is the most common type of epithelial ovarian cancer.
Notes
Author Contributions
Conceived and designed the analysis: Cho HY, Kim YB, No JH.
Collected the data: Cho HY, Kim YB, Park WH.
Contributed data or analysis tools: Kim YB.
Performed the analysis: Cho HY, Kim YB, Park WH, No JH.
Wrote the paper: Cho HY, Park WH.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgements
This study was supported by grant no. 02-2018-003 from the Seoul National University Bundang Hospital Research Fund.