Differing Outcomes of Patients with High Hyperdiploidy and ETV6-RUNX1 Rearrangement in Korean Pediatric Precursor B Cell Acute Lymphoblastic Leukemia
Article information
Abstract
Purpose:
Recent cooperative trials in pediatric acute lymphoblastic leukemia (ALL) report long-term event-free survival (EFS) of greater than 80%. In this study, we analyzed the outcome and prognostic factors for patients with precursor B cell ALL (n=405) diagnosed during a 10-year period (2005–2015) at our institution.
Materials and Methods
All patients were treated with a uniform institutional regimen based on four risk groups, except for steroid type; patients diagnosed up till 2008 receiving dexamethasone, while subsequent patients received prednisolone. None of the patients received cranial irradiation in first complete remission.
Results
The 10-year EFS and overall survival was 76.3%±2.3% and 85.1%±1.9%. Ten-year cumulative incidence of relapse, any central nervous system (CNS) relapse and isolated CNS relapse was 20.8%±2.2%, 3.7%±1.1%, and 2.5%±0.9%, respectively. A comparison of established, good prognosis genetic abnormalities showed that patients with high hyperdiploidy had significantly better EFS than those with ETV6-RUNX1 rearrangement (10-year EFS of 91.2%±3.0% vs. 79.5%±4.4%, p=0.033). For the overall cohort, male sex, infant ALL, initial CNS involvement, and Philadelphia chromosome (+) ALL were significant factors for lower EFS in multivariate study, while high hyperdiploidy conferred favorable outcome. For high and very high risk patients (n=231), high hyperdiploidy was the only significant factor for EFS in multivariate study.
Conclusion
Regarding good prognosis genetic abnormalities, patients with high hyperdiploidy had significantly better outcome than ETV6-RUNX1(+) patients. High hyperdiploidy was a major, favorable prognostic factor in the overall patient group, as well as the subgroup of patients with higher risk.
Introduction
Recent multi-center and multi-national cooperative trials in pediatric acute lymphoblastic leukemia (ALL) report long-term event-free survival (EFS) rates of > 80% and overall survival (OS) rates of ≥ 90% [1–3]. Regarding the overall treatment strategy for pediatric ALL, the addition of an intensification or reinduction phase for all patients regardless of risk group contributed to better outcome [4–6]. For central nervous system (CNS) prophylaxis, limiting cranial irradiation to high risk precursor B cell (Pre-B) and T-cell ALL patients at a dose of 12 Gy was found to be a viable option to minimize long-term neurologic sequelae [7]. Other cooperative studies eliminated cranial irradiation in all patients while undertaking CNS prophylaxis through systemic and intrathecal chemotherapy, and showed low rates of CNS relapse [8,9].
Treatment according to risk group is a cardinal feature of contemporary pediatric ALL therapy, with the designation of risk group based on genetic features of the leukemic blast and response to early therapy, as measured by minimal residual disease [10,11]. Genetic abnormalities significantly influence outcome, with patients with high hyperdiploidy or ETV6-RUNX1 rearrangement known to have favorable prognosis [12].
Previously, we reported the outcome for 295 pediatric ALL patients treated with our institutional regimen, key features of which included intensification therapy based on risk, and the elimination of cranial irradiation in all patients in first complete remission (CR) [13]. Important prognostic variables in this study were high hyperdiploidy as a predictor of favorable outcome, while infant ALL had significantly worse survival. In this study, we undertook a follow-up analysis based on a larger number of patients than the original cohort, and limited to Pre-B ALL patients, to evaluate both rates of long-term survival, and significant predictors of outcome in Pre-B ALL.
Materials and Methods
1. Study group
Patients diagnosed with Pre-B ALL during a 10-year period from January 2005 to June 2015 at the Department of Pediatrics, The Catholic University of Korea were included in the study (n=405).
2. Risk group classification and treatment strategy
Key features of our risk group classification scheme and chemotherapy, which consisted of a uniform institutional regimen, were previously described [13]. Risk group classification was done after remission induction chemotherapy according to the following criteria for Pre-B ALL:
- Low risk (all of the following criteria): age ≥ 1 and < 10 years old, initial white blood cell (WBC) count < 50×109/L, trisomies of 4, 10 and 17 or ETV6-RUNX1(+)
- Standard risk: as for low risk, except for lack of trisomies of 4, 10 and 17 or ETV6-RUNX1
- High risk (any one of the following criteria, irrespective of low risk cytogenetic features): age ≥ 10 and < 15 years old, initial WBC count ≥ 50×109/L and < 100×109/L, initial CNS or testicular involvement, poor prephase steroid response, E2A-PBX1(+), KMT2A rearrangement(+), minimal residual disease (MRD)(+) at end of remission induction (as measured by reverse transcription polymerase chain reaction [RT-PCR] or real-time quantitative [RQ-PCR] for patients with recurrent genetic abnormalities)
- Very high risk (any one of the following, irrespective of low risk cytogenetic features): age ≥ 15 years old, initial WBC count ≥ 100×109/L, Philadelphia chromosome-positive (Ph(+)) ALL, infant ALL, hypodiploidy (≤ 44 chromosomes), induction failure
The major features of our treatment strategy included the following:
- Four drug remission induction for all patients (vincristine, steroid, l-asparaginase, one dose of daunorubicin on day 1), preceded by 1 week of prephase steroid therapy (S1 Table).
- Intensification therapy based on patient risk, with low and standard risk patients receiving one phase of intensification, while high and very high risk patients received two phases of treatment (S2–S5 Tables).
- Maintenance therapy of 96 weeks for all patients regardless of sex. Treatment included pulses of vincristine and steroid every 4 weeks, as well as daily 6-mercaptopurine and weekly methotrexate (MTX). Of note, patients received high dose MTX (3 g/m2) and intrathecal MTX every 12 weeks during maintenance therapy (S6 Table).
- Omission of cranial irradiation in all patients. Treatment of CNS3 consisted of multiple triple intrathecal infusions (methotrexate, cytarabine, hydrocortisone) during remission induction, and use of dexamethasone in all phases of steroid treatment.
- Allogeneic hematopoietic cell transplantation (HCT) in first CR for patients with Ph(+) ALL, hypodiploidy, and induction failure. Since September 2011, infant ALL with KMT2A rearrangement was also included as an indication for allogeneic HCT in first CR.
- For Ph(+) ALL, imatinib was given concurrent with chemotherapy from consolidation up till HCT, as reported previously [14]. Imatinib was not given after HCT, unless the patient showed an increase in RQ-PCR value for BCR-ABL1.
Patients diagnosed from 2005–2008 received dexamethasone during all phases of steroid treatment, while patients diagnosed from 2009 onwards received prednisolone.
3. Study objectives
Primary objectives of the study were to determine the long-term EFS and OS of our study group, as well as long-term incidence of overall relapse, CNS relapse, isolated CNS relapse, and nonrelapse mortality (NRM) for patients who achieved CR. We also aimed to identify key prognostic factors for EFS through both univariate and multivariate study for the entire study group, as well as for two subgroups based on risk (low and standard/high and very high).
4. Statistical analysis
EFS was determined from time of diagnosis to last follow-up in CR or first event, which included relapse, death, or primary refractory status. Patients who died during remission induction or due to primary refractory disease were considered to have events at time zero. OS was determined from date of diagnosis to last follow-up or death. Survival estimates were calculated by the Kaplan-Meier method, and univariate study of risk factors for EFS was done with the log-rank test. Multivariate analysis was done by Cox proportional hazard regression for variables found significant in univariate study. For patients who achieved CR, rate of relapse and NRM was determined by the cumulative incidence function. Patient follow-up was done up till June 30, 2019. p < 0.05 was considered significant.
Results
1. Clinical and genetic characteristics, response to initial therapy
Most of the patients received prednisolone (n=259, 64%), rather than dexamethasone during treatment (Table 1). Regarding genetic features of the leukemic blast, ETV6-RUNX1 rearrangement was most common, followed by high hyperdiploidy, comprising 93 (23%) and 91 (23%) patients, respectively (Table 2). After 4-drug remission induction chemotherapy, 392 patients (97%) achieved CR, nine patients (2%) failed to achieve CR, and four patients (1%) died during remission induction. Overall risk group classification was as follows: low risk, 94 (23%); standard risk, 80 (20%); high risk, 114 (28%); and very high risk 117 (29%).
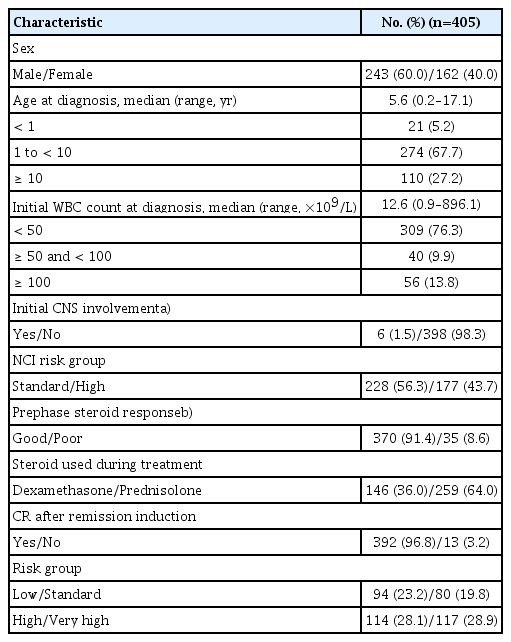
Patient characteristics
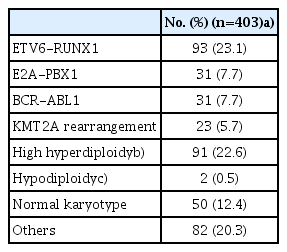
Genetic abnormalities of the patients
2. Survival and events
Forty-two patients (10%) received allogeneic HCT in first CR. With a median follow-up duration of 91.2 months (range, 0.7 to 169.6 months), the 10-year EFS and OS was 76.3%±2.3% (314/405) and 85.1%±1.9% (348/405), respectively (Fig. 1A and B). Events included the following: relapse, 78 (19%); death in CR, nine (2%); and death during remission induction, four (1%) (Table 3). Ten-year cumulative incidence of relapse, any CNS relapse and isolated CNS relapse was 20.8%±2.2%, 3.7%±1.1%, and 2.5%±0.9%, respectively. For patients who achieved CR, 10-year cumulative incidence of NRM was 2.2%±0.8%. Of the nine patients with NRM, eight died from causes related to allogeneic HCT.
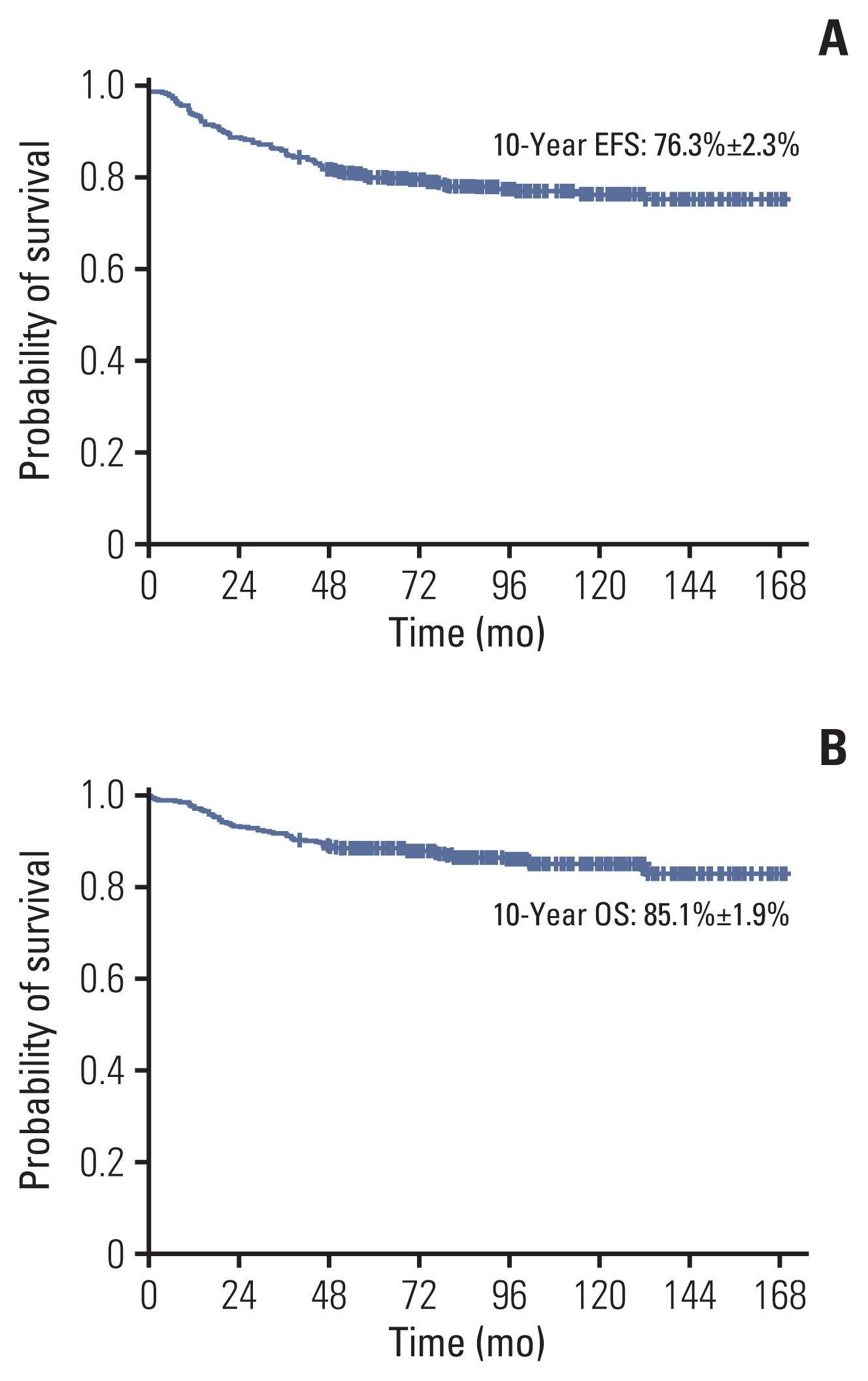
(A) The 10-year event-free survival (EFS) of overall cohort. (B) The 10-year overall survival (OS) of overall cohort.

Events in the overall cohort
3. Comparison of low risk genetic abnormalities
A comparison of EFS of patients with ETV6-RUNX1 and high hyperdiploidy showed that patients with high hyperdiploidy had significantly better outcome (10-year EFS of 91.2%±3.0% [83/91] vs. 79.5%±4.4% [75/93], p=0.033) (Fig. 2A). For patients with high hyperdiploidy, 10-year EFS rates for those with overall low, standard risk and those with overall high, very high risk showed comparable favorable outcomes (92.9%±3.4% for patients with low, standard risk [53/57] vs. 88.2%±5.5% for patients with high, very high risk [30/34], p=0.437) (Fig. 2B). In contrast, patients with ETV6-RUNX1(+) ALL in the higher overall risk groups had significantly worse EFS than those in the lower overall risk groups (10-year EFS of 86.1%±4.3% for patients with low, standard risk [58/67] vs. 61.5%±10.8% for patients with high, very high risk [17/26], p=0.014) (Fig. 2C).
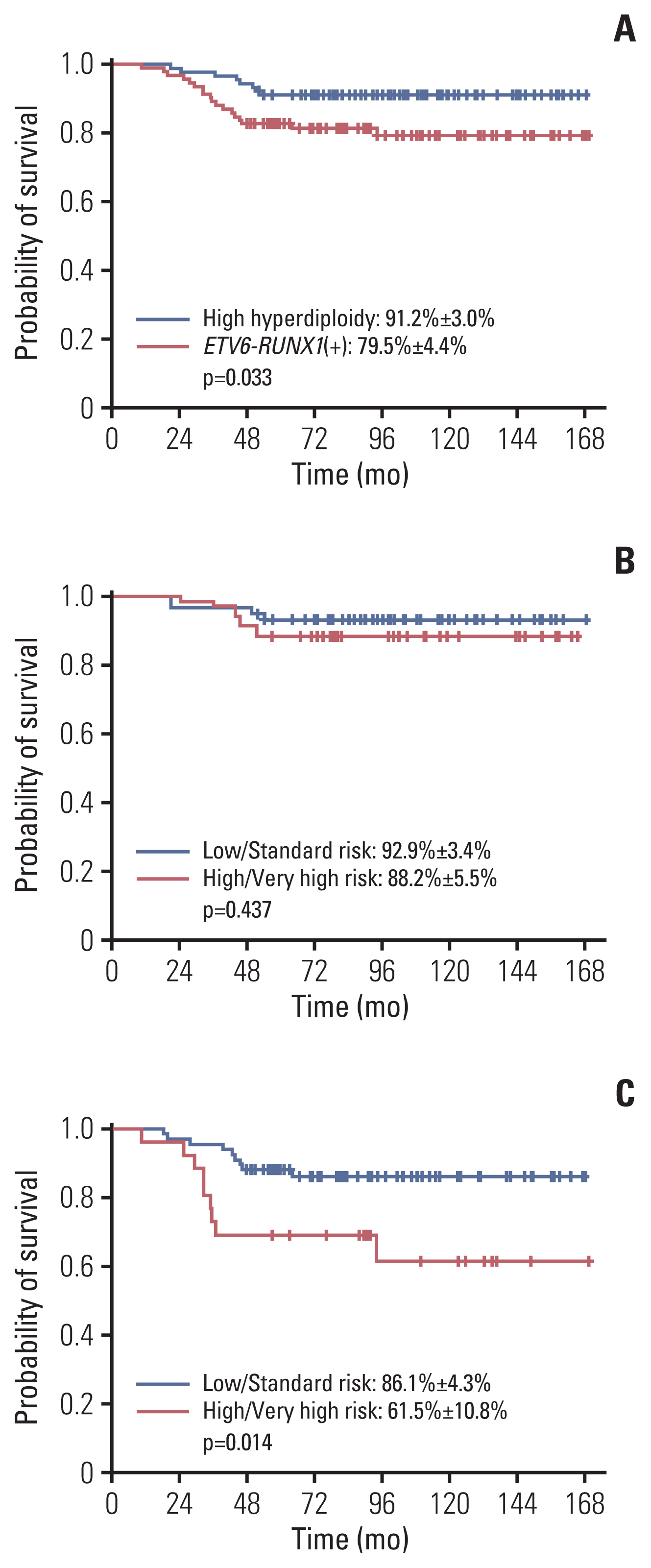
(A) Comparison of 10-year event-free survival (EFS) of patients with high hyperdiploidy (91.2%±3.0%) and ETV6-RUNX1 rearrangement (79.5%±4.4%). (B) The 10-year EFS of high hyperdiploidy patients according to risk group: low and standard risk 92.9%±3.4%; high and very high risk 88.2%±5.5%. (C) The 10-year EFS of ETV6-RUNX1(+) patients according to risk group: low and standard risk 86.1%±4.3%; high and very high risk 61.5%±10.8%.
4. Prognostic factors
Patient sex, age at diagnosis, initial WBC count, CNS involvement at diagnosis, response to prephase steroid, and type of genetic abnormality of the leukemic blast proved to be significant predictors of EFS (Table 4). Also, patients treated with dexamethasone had significantly better EFS than those treated with prednisolone (10-year EFS 82.1%±3.2% vs. 70.3%±4.3%, p=0.048). In multivariate study, male sex, infant ALL, initial CNS involvement, and Ph(+) ALL were significant factors for lower EFS, while high hyperdiploidy conferred favorable outcome (Table 5).
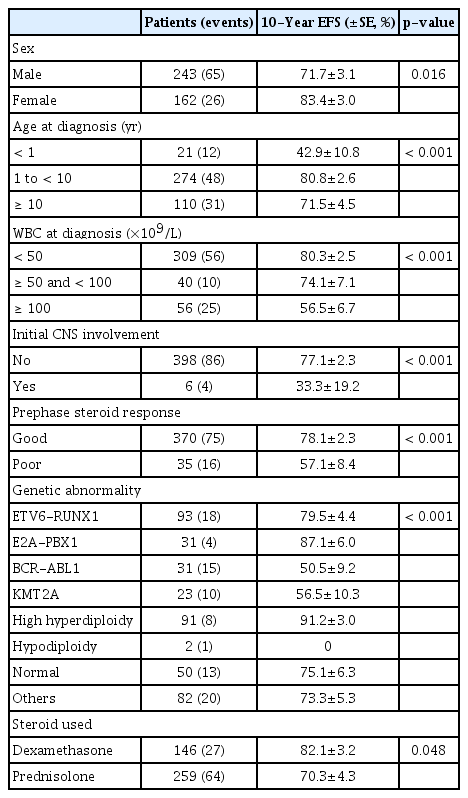
Risk factors for EFS (n=405)
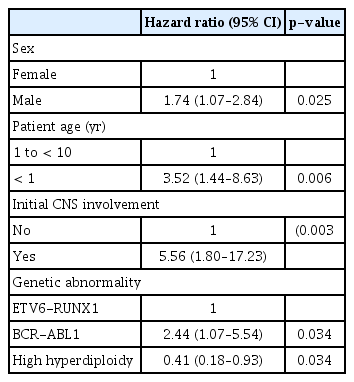
Multivariate study of risk factors for EFS
Results of treatment according to patient subgroups were as follows.
1) Patient sex
The 10-year EFS for boys was significantly inferior to that for girls (71.7%±3.1% vs. 83.4%±3.0, p=0.016) (Table 4). For the 52 boys who were initially treated with chemotherapy only in first CR and subsequently relapsed, the median time to relapse was 30.8 months from diagnosis (range, 3.5 to 113.6 months). Twenty-one patients (40%) relapsed after treatment completion, 21 (40%) during maintenance therapy, while the remaining 10 patients (20%) relapsed early, prior to start of maintenance therapy.
2) Infant ALL
The 10-year EFS for the infants (n=21) was 42.9±10.8, significantly inferior to other age groups (Table 4). According to presence of KMT2A rearrangement, the 10-year EFS was 50.0%±17.7% for the eight patients without KMT2A rearrangement, none of whom received allogeneic HCT in first CR, and 38.5%±13.5% for the 13 patients with KMT2A rearrangement (KMT2A(+)), six of whom received transplantation in first CR (p=0.708). Outcome for KMT2A(+) patients according to allogeneic HCT status was as follows: 10-year EFS of 50.0%±20.4% (3/6) for those who received HCT in first CR, and 28.6%±17.1% (2/7) for those who initially received chemotherapy only (p=0.158).
3) Initial CNS leukemia
Of the six patients with CNS leukemia at diagnosis, four experienced an event (10-year EFS, 33.3%±19.2%), including two patients with infection-related death during remission induction, one patient with bone marrow relapse, and one patient with isolated CNS relapse.
4) Ph(+) ALL
Twenty-five of the 31 patients with Ph(+) ALL (81%) underwent allogeneic HCT in first CR; six patients did not undergo HCT due to lack of human leukocyte antigen fully-matched hematopoietic cell donor, early death, or comorbidities. Of the 25 patients who underwent first CR HCT, four patients relapsed and six patients died of NRM (10-year EFS, 59.1%±10.0%); of the six patients who did not receive HCT, three patients relapsed, and two patients died of NRM (10-year EFS, 16.7%±15.2%, p=0.057 when comparing the two groups). Ten-year OS of the Ph(+) ALL subgroup was 65.7%±8.9% (21/31).
5. Outcome and prognostic factors according to risk group
The 10-year OS according to risk group was as follows: low risk, 95.7%±2.1% (90/94); standard risk, 98.8%±1.2% (79/80); high risk, 84.4%±3.7% (97/114); and very high risk, 68.7%±4.6% (82/117) (Fig. 3). For the 174 patients with low and standard risk ALL, male sex (10-year EFS of 85.1%±3.7% [84/98] vs. 96.0%±2.3% for girls [73/76], p=0.024), and treatment with prednisolone (10-year EFS of 85.6%±3.5% [98/113] vs. 96.7%±2.3% [59/61] for treatment with dexamethasone, p=0.030) predicted worse outcome in univariate study. None of the factors proved significant in multivariate analysis.
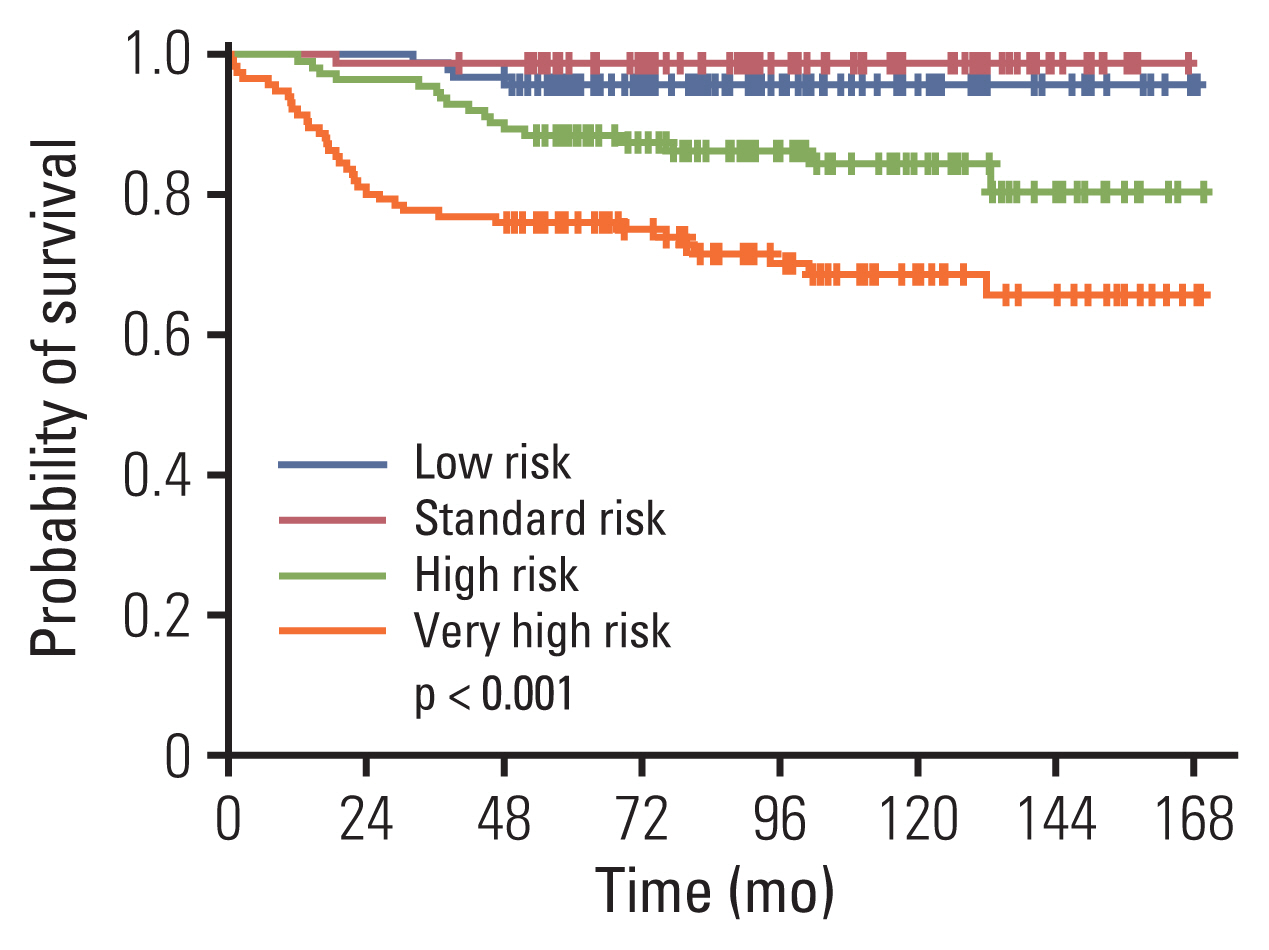
The 10-year overall survival of patients according to overall risk group: low risk, 95.7%±2.1%; standard risk, 98.8%±1.2%; high risk, 84.4%±3.7%, very high risk, 68.7%±4.6%.
Regarding the 231 patients with high and very high risk ALL, patient age at diagnosis, initial WBC count, CNS involvement, prephase steroid response, and the type of genetic abnormality had a significant impact on EFS in univariate study (S7 Table). In multivariate study, high hyperdiploidy was the only significant prognostic factor, with patients harboring this genetic abnormality having improved EFS (hazard ratio, 0.26; 95% confidence interval, 0.08 to 0.85; p=0.026).
Discussion
In this study, we report the outcome and prognostic factors of 405 pediatric Pre-B ALL patients diagnosed and treated with a uniform treatment regimen at our institution for a period of ten years. In multivariate study, we found that male sex, infant ALL, initial CNS involvement and BCR-ABL1 rearrangement predicted significantly worse EFS, while patients with high hyperdiploidy had favorable outcome.
Although one past nationwide study showed a significantly higher relapse-free survival for girls compared with boys [15], recent studies have shown comparable outcome according to patient sex [2,9,10]. In our study, boys had lower 10-year EFS compared with girls, and patient sex proved to be a significant prognostic factor in multivariate analysis. Considering the potential for inferior outcome for boys, several clinical trials administered differing chemotherapy regimens according to patient sex, specifically by lengthening the duration of maintenance chemotherapy for boys compared with girls [3,6,16]. In our patient group, a significant proportion of the boys who were initially treated with chemotherapy only relapsed after the end of therapy (21 of 52 patients). Increasing the duration of maintenance therapy holds the possibility of preventing relapse for some of these patients.
Outcome for infant ALL patients was poor regardless of KMT2A rearrangement status. Previous studies reported better survival for KMT2A germline infant ALL patients, with long-term EFS ranging from 76% to 96% [17,18]. Considering the small number of KMT2A germline infant ALL patients in our study (n=8), further analysis based on a larger number of patients is necessary to evaluate the outcome of KMT2A germline patients treated with very high risk chemotherapy. Survival for KMT2A(+) infant ALL was poor despite treatment with allogeneic HCT in first CR in some of these patients, consistent with the low survival rate reported in past studies [18,19]. Novel treatment strategies may improve the survival of infant ALL, as shown in a recent study which reported outcomes for relapsed or refractory KMT2A(+) infant ALL treated with the bi-specific T-cell engager blinatumomab as a bridge to transplant [20].
Cranial irradiation has remained an important component of therapy for high risk ALL, either in the prophylactic setting, or for treatment of initial CNS involvement [3,11,21]. Recent cooperative trials, however, have shown that cranial irradiation may be replaced by intensive intrathecal chemotherapy, resulting in low incidence of CNS relapse [8–10]. In our study, none of the patients received cranial irradiation in first CR; the subsequent 10-year cumulative incidence of CNS relapse of 3.7% is similar to rates reported by recent trials in which cranial irradiation was incorporated as prophylaxis, or for the treatment of CNS involvement [3,11]. Only six patients in our study had CNS involvement at diagnosis (CNS3). Considering that two of the four events experienced by these patients were infection-related deaths during remission induction, rather than relapse, evidence from our data is limited regarding potential need for therapeutic cranial irradiation in this small subset of patients.
Recent studies have shown that Ph(+) ALL patients treated with conventional chemotherapy combined with tyrosine kinase inhibitors (TKIs), such as imatinib or dasatinib, have outcome similar to historical cohorts treated with allogeneic HCT [22,23]. Our treatment strategy for Ph(+) ALL patients has been to undertake early HCT in first CR. Of six Ph(+) ALL patients who did not receive HCT in first CR but continued treatment with chemotherapy and imatinib, one survives event-free while three patients relapsed and two patients died of NRM. Considering that of the 25 patients who received HCT in first CR, four patients relapsed and six patients died of NRM post-HCT, attempts to lower both relapse and transplant-related toxicity are necessary to improve the outcome of HCT for Ph(+) ALL patients. Our previous analysis of Ph(+) ALL patients showed that post-consolidation MRD was the only significant factor predicting HCT outcome [14]. Combination of chemotherapy and uninterrupted TKI treatment prior to HCT with the aim of minimizing pre-transplant MRD may improve the results of transplant for Ph(+) ALL patients.
Good risk genetic features of Pre-B ALL include ETV6-RUNX1 translocation and high hyperdiploidy, defined as 51–65 chromosomes per cell. Past studies show that the excellent survival rate of high hyperdiploidy may depend on other factors such as the age of the patient and the presence of specific chromosomal trisomies [24,25]. Patients with high hyperdipoidy classified and treated in a higher overall risk group may have inferior survival compared with high hyperdiploidy patients in lower risk groups [26]. In our study, 83 of 91 patients with high hyperdiploidy survive event-free, resulting in long-term EFS of 91%. Importantly, a risk factor analysis limited to high and very high risk patients showed that the presence of high hyperdiploidy maintained significance as a favorable prognostic factor in multivariate analysis, with no difference in outcome between high hyperdiploidy patients in the low and standard risk groups and those in the higher risk groups within the context of risk group-based chemotherapy. Hence, in our study, the favorable effect of high hyperdiploidy was not modified by other adverse variables.
ETV6-RUNX1(+) ALL may be more heterogeneous in terms of prognosis, with outcome influenced by variables such as MRD, and concurrent genetic abnormalities [27–29]. In our study, ETV6-RUNX1(+) ALL patients had significantly different outcomes according to overall risk group. Greater prognostic heterogeneity of ETV6-RUNX1(+) ALL patients may be the key factor for patients with high hyperdiploidy having significantly better EFS than those with ETV6-RUNX1 fusion in our study group. Overall, our results underscore the disparate outcome for the two canonical good prognosis genetic abnormalities of Pre-B ALL, and lower survival of our ETV6-RUNX1(+) ALL patients (10-year EFS of 79.5±4.4%) compared to the excellent survival rates reported in past studies [2,8,9].
A clear limitation of our study was the lack of implementation of common MRD testing in our patients, with MRD done by RT- or RQ-PCR only for patients with recurrent genetic abnormalities, and the subsequent inability to confirm the well-known role of MRD as a prognostic factor. We also emphasize that due to our risk group stratification most of our patients were high or very high risk, in contrast to reported clinical trials in which most of the ALL patients were standard risk [6,8,10].
In summary, through an analysis of 405 Pre-B ALL patients treated over a period of more than 10 years, we found a long-term OS of 85%. Important negative prognostic factors included male sex, infant ALL, initial CNS involvement, and BCR-ABL1 rearrangement, while patients with high hyperdiploidy uniformly had excellent outcome regardless of overall risk group. Regarding established good prognosis genetic abnormalities, patients with high hyperdiploidy had significantly better outcome than patients with ETV6-RUNX1 rearrangement. Therapy modification tailored to poor prognosis factors may further improve survival of our Pre-B ALL patients.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
The study received institutional review board (IRB) approval (number KC19RESI0504). As the study was retrospective, the need for informed consent from the patients was waived by the IRB.
Author Contributions
Conceived and designed the analysis: Lee JW, Kim S, Jang PS, Chung NG, Cho B.
Collected the data: Lee JW.
Performed the analysis: Lee JW.
Wrote the paper: Lee JW, Kim S, Jang PS, Chung NG, Cho B.
Conflicts of Interest
Conflicts of interest relevant to this article was not reported.
Acknowledgements
We express heartfelt gratitude to the nursing staff associated with the Division of Hematology/Oncology, Department of Pediatrics, The Catholic University of Korea, and, above all, to the patients and their families.