Prevalence of PALB2 Germline Mutations in Early-onset and Familial Breast/Ovarian Cancer Patients from Pakistan
Article information
Abstract
Purpose
Partner and localizer of BRCA2 (PALB2) is a breast cancer susceptibility gene that plays an important role in DNA repair. This is the first study assessing the prevalence of PALB2 mutations in early-onset and familial breast/ovarian cancer patients from Pakistan.
Materials and Methods
PALB2 mutation screening was performed in 370 Pakistani patients with early-onset and familial breast/ovarian cancer, who were negative for BRCA1, BRCA2, TP53, CHEK2, and RAD51C mutations, using denaturing high-performance liquid chromatography analysis. Mutations were confirmed by DNA sequencing. Novel PALB2 alterations were analyzed for their potential effect on protein function or splicing using various in silico prediction tools. Three-hundred and seventy-two healthy controls were screened for the presence of the identified (potentially) functional mutations.
Results
A novel nonsense mutation, p.Y743*, was identified in one familial breast cancer patient (1/127, 0.8%). Besides, four in silico-predicted potentially functional mutations including three missense mutations and one 5' untranslated region mutation were identified: p.D498Y, novel p.G644R, novel p.E744K, and novel c.-134_-133delTCinsGGGT. The mutations p.Y743* and p.D498Y were identified in two familial patients diagnosed with unilateral or synchronous bilateral breast cancer at the ages of 29 and 39, respectively. The other mutations were identified in an early-onset (≤ 30 years of age) breast cancer patient each. All five mutations were absent in 372 healthy controls suggesting that they are disease associated.
Conclusion
Our findings show that PALB2 mutations account for a small proportion of early-onset and hereditary breast/ovarian cancer cases in Pakistan.
Introduction
Breast cancer has a substantial impact on the overall tumor burden in Pakistan comprising 40% of all female malignancies. In Pakistan, monoallelic germline mutations in the high- and moderate-penetrance breast cancer susceptibility genes BRCA1, BRCA2, TP53, CHEK2, and RAD51C account for approximately 25% of early-onset and familial breast cancer suggesting that other susceptibility gene(s) may be involved. Recently identified PALB2 gene, a partner and localizer of BRCA2, acts as a link between BRCA2 and BRCA1 and enables DNA repair [1]. Biallelic mutations in PALB2 (FANCN) cause Fanconi anemia, with clinical features similar to those caused by biallelic mutations in BRCA2 (FANCD1) [2]. Monoallelic mutations in PALB2 confer susceptibility to breast cancer suggesting that PALB2 is another candidate to be a breast cancer susceptibility gene [3]. Deleterious PALB2 mutations are estimated to confer a 35% lifetime risk of breast cancer for the carriers [4].
PALB2 mutation screening in BRCA1/2-negative early-onset or familial breast cancer patients performed previously in different populations have yielded variable results. In a British study, monoallelic mutations in PALB2 were identified in 1.1% of familial breast cancer patients (10/923) and were absent in controls (0/1,084) [3]. In a study conducted in Finland, a recurrent PALB2 mutation, c.1592delT, was identified in 2.7% of familial breast cancer patients (3/113) and in 0.2% of controls (6/2,501) [5]. Mutations were identified in families from Europe, North America, and Australia at frequencies ranging from 0.3% to 5.5% [6-9], while mutations were absent in families from Canada [10], United States [11], and Chile [12].
In Asia, little is known about the contribution of PALB2 mutations to early-onset and familial breast/ovarian cancer. Deleterious PALB2 mutations were reported in families from China, Korea, and Malaysia/Singapore with frequencies varying from 0.8% to 1.6% [13-15] and were absent in Japanese families [16]. Given the limited data on genetic variability of PALB2 in South Asia and the fact that only 25% of hereditary breast/ovarian cancer in Pakistan is attributed to germline mutations in BRCA1/2, TP53, CHEK2, and RAD51C [17-20], we assessed and report the prevalence of PALB2 mutations in 370 early-onset and familial breast/ovarian cancer patients from this population, who had tested negative for mutations in these five breast cancer susceptibility genes. Functional and potentially functional mutations were screened in 372 healthy controls.
Materials and Methods
1. Study population
The study included 370 early-onset and familial breast/ovarian cancer patients who were diagnosed with invasive breast cancer or epithelial ovarian cancer. The patients were evaluated and accrued to the study at the Shaukat Khanum Memorial Cancer Hospital and Research Center (SKMCH&RC) in Lahore, Pakistan, from June 2001 to January 2012. Patients were classified into six groups based on the age at the time of disease onset or family history of breast/ovarian cancer: A1, families with one female breast cancer diagnosed ≤ 30 years of age; A2, families with two first- or second-degree (through a male) female relatives diagnosed with breast cancer, at least one diagnosed ≤ 50 years of age; A3, families with at least three cases of breast cancer, at least one diagnosed ≤ 50 years of age; A4, families with one male breast cancer case diagnosed at any age; B, families with at least one female breast cancer and one ovarian cancer at any age, and C1, families with at least one ovarian cancer diagnosed ≤ 45 years of age. All index patients were previously tested and shown to be negative for deleterious mutations in the BRCA1/2, CHEK2, and RAD51C genes [17,19,20]. Of these index patients, 290 cases were also tested negative for disease-causative mutations in the TP53 gene [18]. Patients with bilateral breast cancer or breast and ovarian cancer were considered as two independent primary cancers. A description of the study participants is shown in Table 1.
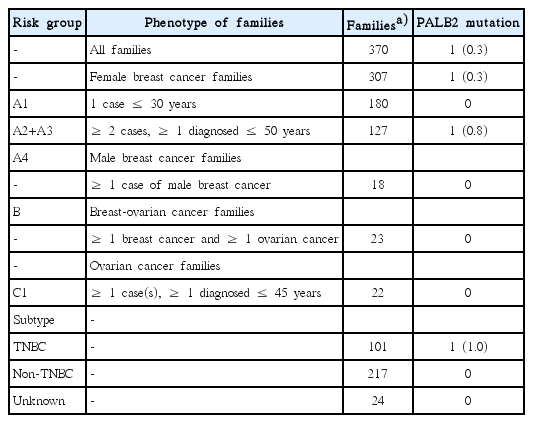
Frequency of PALB2 mutations according to family structure and TNBC subtype
Immunohistochemical analyses of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression were performed on the breast tumor of the index patients as described previously [21].
The control group comprised of 372 healthy Pakistani women. Their recruitment details have been previously reported [22].
2. Molecular analyses
Genomic DNA extraction was performed as described previously [22]. The complete coding sequence and exon-intron junctions of the PALB2 gene (GenBank accession number NM_024675.3) were screened in the 370 index patients by denaturing high-performance liquid chromatography (DHPLC) analysis using WAVE 4500 DNA Fragment Analysis System (Transgenomics, Omaha, NE). Custom designed polymerase chain reaction (PCR)-primer pairs were used (Transgenomic Personalized Customer Support). DHPLC is a temperature modulated heteroduplex analysis which relies upon the physical changes in DNA molecules induced by mismatch heteroduplex formation [23]. Heteroduplexes were formed by denaturing the PCR product. The amplified product was loaded on a unique DNA separation matrix (Transgenomics) with homoduplexes and heteroduplexes eluting to it differentially under denaturing conditions. The elution profiles of heterduplexes were easily distinguished from those of homoduplexes. This technique has also been reported previously to detect PALB2 mutations [11,13,24,25]. When available, a mutation positive control for each exon was included in each analysis. Primer sequences, the setup of PCR reactions, cycling conditions, and DHPLC running conditions are available upon request.
Samples revealing variant DHPLC profiles were bi-directionally sequenced using an automated 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA). The presence of identified pathogenic and in silico-predicted potentially deleterious PALB2 mutations was assessed in 372 controls.
3. In silico analyses
All identified novel PALB2 missense variants were analyzed for their potential effect on protein function by using five different computational algorithms, Align-GVGD, SIFT, MutationTaster, Polyphen-2, and SNAP2 in the default settings. In brief, Align-GVGD is based on the biophysical features of amino acids and multiple sequence alignments of protein (http://agvgd.iarc.fr/agvgdinput.php). SIFT predicts the evolutionary conservation of amino acids within protein families (http://sift.jcvi.org). MutationTaster prediction is based on regulatory features such as histone modification sites or transcription factor binding sites, evolutionary conservation of amino acid or nucleotide, and splice site predictions (http://www.mutationtaster.org/). PolyPhen-2 predicts the influence of an amino acid substitution on the structure and function of human protein (http://genetics.bwh.harvard.edu/pph2/). SNAP2 is based on multiple sequence alignment and predict changes on the secondary structure and compare the solvent accessibility for mutant and wild type proteins (http://www.rostlab.org/services/snap/submit) [26].
Novel intronic variants were also analyzed for their potential effect on splicing using the splice prediction algorithms, SpliceSiteFinder-like (http://www.umd.be/searchSpliceSite.html), MaxEntScan (http://genes.mit.edu/burgelab/maxent/), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), GeneSplicer (http://ccb.jhu.edu/software/genesplicer/), and HumanSplice Finder (http://www.umd.be/HSF/) by means of the Alamut software interface (Interactive Biosoftware) in default settings. All these algorithms compare the splice site signal of wild type sequence with that of the mutated sequence. Moreover, one novel variant identified in 5′ untranslated region (5′ UTR) of PALB2 was analyzed to predict its potential effect on transcription factor binding sites using PROMO (v3.0.2) on-line program (http://alggen.lsi.upc.es/cgibin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3).
4. Ethical statement
Informed written consent was obtained from all study participants prior to providing a blood sample. The study was approved by the Institutional Review Board (IRB) of the SKMCH&RC (IRB approval number ONC-BRCA-001/2).
Results
The present study included 370 early-onset and familial breast/ovarian cancer patients from Pakistan, who were negative for BRCA1/2, TP53, CHEK2, and RAD51C mutations. Of these, 180 were diagnosed with early-onset breast cancer (≤ 30 years of age), 127 belonged to families with two or more breast cancers, 23 to families with both breast and ovarian cancer, 22 to families with at least one ovarian cancer (diagnosed ≤ 45 years of age), and 18 to families with male breast cancer (Table 1). The median age of the disease onset was 30 years (range, 19 to 73 years) for female breast cancer (n=324), 47.5 years (range, 30 to 73 years) for male breast cancer (n=18), and 33 years (range, 22 to 60 years) for ovarian cancer (n=35). One-hundred and one index patients presented with triple-negative breast cancer (TNBC) and 217 with non-TNBC. The median age of the disease onset was 28 years (range, 19 to 67 years) for patients with TNBC and 30 years (range, 19 to 73 years) for those with non-TNBC.
Overall, 31 different heterozygous PALB2 variants were detected. Of these, 13 were novel: one nonsense mutation, four missense variants, one silent variant, six intronic variants, and one 5′ UTR variant (Table 2). The remaining 18 variants have been previously reported in other populations: eight missense variants, four silent variants, five intronic variants, and one 5′ UTR variant.

PALB2 germline mutations in early-onset and familial breast/ovarian cancer patients from Pakistan
The novel nonsense mutation at nucleotide position 2229 in exon 5, c.2229T>A (p.Y743*), was detected in a 29-year-old breast cancer patient of Punjabi ethnicity. The patient presented with a grade 3, invasive ductal carcinoma (IDC) of TNBC phenotype. A sister of the index patient was diagnosed with breast cancer at the age of 41 years.
The remaining 12 novel variants were analyzed for their potential functional effect by in silico analysis. Two missense variants p.G644R and p.E744K and the previously reported missense variant p.D498Y [14,16,27] were predicted to be potentially deleterious by three out of five in silico analysis tools (Table 3). The 5′ UTR variant c.-134_-133-del-TCinsGGGT was predicted to create a splice donor site by three out of five splice-site prediction tools implying that it was disease associated (Table 4). This variant was also predicted to create the binding sites for two transcription factors, GR-alpha (T00337) and RXR-alpha (T01345), using PROMO (v3.0.2) on-line program.

In silico analyses of PALB2 coding variants identified in early-onset and familial breast/ovarian cancer patients from Pakistan

In silico analyses of PALB2 noncoding variants identified in early-onset and familial breast/ovarian cancer patients from Pakistan
The potentially disease-causative missense mutations p.G644R and p.E744K were identified in two women of Punjabi or Pathan ethnic group, who were diagnosed with early-onset breast cancer at the ages of 30 years, respectively. Their tumors were grade 3 IDCs, which were, ER-positive, PR-positive, and HER2-negative. The other missense mutation, p.D498Y, was identified in a patient of Pathan origin, who was diagnosed with synchronous bilateral breast cancer at 39 years of age. Left-sided breast tumor was grade 2 IDC and ER-positive, PR-negative, and HER2-negative. Right-sided breast tumor was grade 3 IDC and ER-negative, PR-negative, and HER2-positive. The index patient reported a family history of breast cancer and other cancers. The mother and one sister of the index patient were diagnosed with breast cancer at the ages of 53 and 44 years, respectively. Her father was diagnosed with lung cancer at age 69. One sister and paternal uncle of the index patient were diagnosed to have brain cancer at the ages of 34 and 48 years, respectively.
The 5′ UTR variant, c.-134_-133delTCinsGGGT, was identified in an early-onset breast cancer patient of Pathan ethnic group, who was diagnosed with disease at 26 years of age. Her tumor was a grade 2 IDC of TNBC phenotype. This patient had also been diagnosed with adenocarcinoma of the rectum at 25 years of age.
The nonsense mutation and the four in silico-predicted potentially functional mutations were not identified in 372 controls implying that they were disease-causative (Table 2). The remaining 17 variants (seven missense variants, four silent variants, five intronic variants, and one 5’ UTR variant) were reported previously and classified as polymorphisms (Table 2).
Discussion
In this large study conducted in Pakistan, we assessed for the first time the prevalence of PALB2 germline mutations in 370 early-onset and familial breast/ovarian cancer patients negative for BRCA1/2, TP53, CHEK2, and RAD51C mutations. One novel deleterious and four in silico-predicted potentially functional PALB2 mutations (including three novels) were identified. Our study provides additional data on the contribution of PALB2 mutations to hereditary breast/ovarian cancer in an Asian population from Pakistan.
The novel pathogenic PALB2 mutation (p.Y743*) was identified in 0.8% of families with two or more breast cancers (1/127). Similar mutation frequencies were reported in other Asian breast and/or ovarian cancer families from Saudi Arabia (5/725, 0.7%) [28], China (3/360, 0.8%) [13], and Korea (2/235, 0.8%) [14]. Higher frequencies were reported in studies conducted in India (9/752, 1.2%) [29], Malaysia/Singapore (2/122, 1.6%) [15], China (2/108, 1.8%) [30], and Korea (3/120, 2.5%) [31]. This could be due to the Next-Generation Sequencing approach employed in most of these studies. Furthermore, our results are likely to underestimate the PALB2 mutation frequencies as the recently reported large genomic rearrangements were not screened [29,32]. Mutations were not identified in few studies from Japan [16], Chile [12], and United States [11]. In European and North American studies including more than 100 families from Czech Republic, France, Ireland, Germany, Poland, Italy, Finland, Canada, United States, and Australia the mutation frequencies ranged from 0.3% to 5.5% [5-9,33-38]. Altogether these findings suggest that PALB2 mutations account for a small proportion of hereditary breast/ovarian cancer in most populations including the Pakistani population.
Four in silico-predicted potentially functional PALB2 mutations (p.G644R, p.E744K, p.D498Y, and c.-134_-133del-TCinsGGGT) were also identified in this study. The novel missense mutations p.G644R and p.E744K, each identified in an early-onset breast cancer patient, were located in the highly conserved MRG15-binding domain of PALB2 spanning amino acid residues 611 to 764. They may ablate PALB2 interacting with MRG15, which may result in impaired DNA repair [39] leading to genomic instability and cancer. The p.G644R variant was predicted to be deleterious by SIFT, Align-GVGD and SNAP-2 algorithms, while it was predicted as benign by PolyPhen-2 and MutationTaster. The differential prediction may be due to the underlying algorithm’s differences as SIFT uses evolutionary sequence conservation whereas PolyPhen-2 utilizes protein structure information [40]. Since none of these algorithms is 100% predictive, a consensus prediction is reported to improve the prediction performance [41]. Overall, this variant was considered as deleterious by three of the five protein function algorithms. The missense mutation p.D498Y was identified in a familial breast cancer patient. Due to lack of DNA samples, co-segregation of the mutation with breast cancer could not be studied. Previously, this was also reported as an in silico-predicted potentially disease-causative mutation in families from Korea, Japan, and Australia [14,16,27]. The 5′ UTR variant, c.-134_-133delTCinsGGGT, was detected in a patient with personal history of breast and rectal cancer. It was predicted to result in activation of a cryptic splice site. This variant was also predicted to affect two transcription factor binding sites and may alter the promoter activity of PALB2. None of these mutations were detected in 372 controls further suggesting that they are disease-causative; however, functional analyses of the potentially disease-causative mutations will be required for final mutation classification.
No deleterious PALB2 mutations were identified in early-onset breast cancer patients, families with breast and ovarian cancer, ovarian cancer, or male breast cancer. These findings are in keeping with those previously reported in families from Turkey [42], Finland [43], Japan [16], the Netherlands [44], Spain [32], Italy [45], and Australia [46]. In contrast, PALB2 mutations have also been reported in families from Poland [37], Italy [47], Czech Republic [8], and China [13]. These findings suggest that PALB2 mutations may not substantially confer susceptibility to early-onset breast cancer patients and patients from breast and ovarian cancer, male breast cancer or ovarian cancer families.
In our study, the breast tumors associated with the PALB2 truncating (p.Y743*) and in silico-predicted potentially functional (p.D498Y, p.G644R, and p.E744K) mutations presented with high-grade tumors of IDC histology, which is consistent with previous studies conducted among Asian [13-15], European [24,25,48], and North-American patients [49]. In the Pakistani study, the breast tumor linked with the truncating mutation displayed the TNBC phenotype, in agreement with other studies from Europe, North-America and Australia [50-52]. Breast tumors associated with missense mutations were ER-positive, PR-positive, and HER2-negative. Similar hormone receptor expression patterns were also reported in breast tumors of Asian [14], European [48], and North-American patients [53], who harbored PALB2 truncating mutations. The differential expression of hormone receptors could be due to mutation-specific tumor phenotypes in the studied population.
In summary, we have identified one novel pathogenic and four potentially pathogenic PALB2 mutations, three being novel in 370 Pakistani early-onset and familial breast/ovarian cancer patients, negative for mutations in BRCA1/2, TP53, CHEK2, and RAD51C. The frequency of PALB2 mutations in patients with familial breast cancer was 0.8% (1/127), while no mutations were identified in early-onset breast cancer patients and patients from breast and ovarian cancer, male breast cancer or ovarian cancer families. Our findings suggest a marginal contribution of PALB2 mutations to breast cancer susceptibility in Pakistan.
Notes
Conflict of interest relevant to this article was not reported.
Acknowledgements
We thank the patients and healthy individuals for their participation in this study. We are grateful to Saima Faisal for the recruitment of study participants. We thank Zdenek Kleibl, Melissa C. Southey, and Natalia Bogdanova for providing DNA samples of PALB2 mutation controls and Asim Amin for critical review of the manuscript. This study was funded by the internal fund from Shaukat Khanum Memorial Cancer Hospital and Research Centre, Lahore, Pakistan (grant number ONC-BRCA-001/2).