Effect of Necrosis on the miRNA-mRNA Regulatory Network in CRT-MG Human Astroglioma Cells
Article information
Abstract
Purpose
Glioblastoma multiforme (GBM) is the most common adult primary intracranial tumor. The remarkable features of GBM include central necrosis. MicroRNAs (miRNAs) have been considered as diagnostic/prognostic biomarkers for many cancers, including glioblastoma. However, the effect of necrosis on the miRNA expression profile and predicted miRNA-mRNA regulatory information remain unclear. The purpose of this study is to examine the effect of necrotic cells on the modulation of miRNA and mRNA expression profiles and miRNA-mRNA network in CRT-MG cells.
Materials and Methods
We used human astroglioma cells, CRT-MG, treated with necrotic CRT-MG cells to examine the effect of necrosis on the modulation of miRNA and mRNA by next-generation sequencing. For preparation of necrotic cells, CRT-MGcellswere frozen and thawed through cycle of liquid nitrogen–water bath. The putative miRNA-mRNA regulatory relationshipwas inferred through target information, using miRDB.
Results
The necrotic cells induced dysregulation of 106 miRNAs and 887 mRNAs. Among them, 11 miRNAs that had a negative correlation value of p < 0.05 by the hypergeometric test were screened, and their target mRNAs were analyzed by Gene Ontology enrichment analysis. Using the Kyoto Encyclopedia of Genes and Genomes database, we also found several necrotic cell treatment-activated pathways that were modulated by relevant gene targets of differentially expressed miRNAs.
Conclusion
Our result demonstrated that dysregulation of miRNA and mRNA expression profiles occurs when GBM cells are exposed to necrotic cells, suggesting that several miRNAs may have the potential to be used as biomarkers for predicting GBM progression and pathogenesis.
Introduction
Grade IV astrocytoma, also called glioblastoma multiforme (GBM), is considered the most aggressive and malignant form of glial tumors [1]. The remarkable features of GBM include diffused infiltration into normal brain parenchyma, local invasion, angiogenesis, and the presence of necrotic tissues [2,3]. Results obtained from clinical studies show that necrosis has an overall negative impact on patient survival and is linked to a poor prognosis [4]. Our group has demonstrated that necrosis enhances glioblastoma cell migration and invasion [5]. Despite extensive treatments, including surgery, radiotherapy, chemotherapy, immunotherapy, and symptomatic therapy, the median survival for patients with GBM is 12-15 months [6]. Because of the poor overall patient survival, the search for new molecular targets and prognostic indicators is of particular interest in GBM research.
A microRNA (miRNA) is a noncoding, interfering RNA with a length of approximately 22-25 nucleotides. It is capable of binding to the 3′ untranslated region of its target mRNA, resulting in mRNA cleavage and negative post-transcriptional regulation of target gene expression [7]. The decrease of suppressive miRNAs on oncogenes and the increase of inhibitory miRNAs on tumor suppressors control gene expression and play specific roles in cancer cell predisposition and initiation, proliferation, invasion, angiogenesis, inflammation, and metastasis in breast cancer, prostate cancer, nasopharyngeal carcinoma, and glioblastoma [8-12]. Therefore, miRNAs are considered prognostic indicators and potential alternative therapeutic targets for many cancers, including glioblastoma [13-15].
Dysregulation of miRNAs is frequently observed in patients with GBM [16]. Previous studies have demonstrated that numerous miRNAs play a role in both the tumorigenesis and recurrence of GBM through their modulation of several cancer-related genes, such as those encoding proliferation, invasion, radiosensitivity and chemosensitivity, and angiogenesis [8,17-19]. Although numerous efforts have been devoted to the establishment of miRNA-based prognostic and diagnostic biomarkers for predicting the progression of GBM, the molecular profiling of glioblastomas with or without necrosis has not yet been clearly demonstrated.
In this study, we investigated the effect of necrotic cells on the miRNA and mRNA expression profiles as well as the negative miRNA-mRNA correlations in a human astroglioma cell line, CRT-MG. Using next-generation sequencing (NGS) analysis, we profiled the miRNA and mRNA expression levels in CRT-MG cells with or without necrotic cell treatment. The miRNAs and mRNAs showing significant changes were selected, and integrative miRNA-mRNA network analyses were performed.
Materials and Methods
1. Cells
Human astroglioma CRT-MG cells were maintained in Dulbecco’s modified essential medium (cat. LM001-05, WelGENE Inc., Daegu, Korea) containing 10% fetal bovine serum (Gibco, Grand Island, NY), L-glutamine, 100 U/mL penicillin and 10 μg/mL streptomycin. For preparation of the necrotic cells, CRT-MG cells were subjected to five frozen-thaw cycles of liquid nitrogen-water bath treatment to induce a necrotic state in the cells [20]. CRT-MG cells that were not treated with the necrotic cells were used as controls.
2. RNA extraction and NGS
Total cellular RNA was isolated from cells using an RNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Small RNA sequencing was done on the Illumina Hiseq 2500 platform (Illumina, San Diego, CA), using 51-bp single-end sequencing with a fragment size of 145-160 bp. The TruSeq small RNA Library Prep kit (Illumina) was used for preparing the library. To detect known and novel miRNAs, unique clustered reads were aligned with the reference genome as well as with precursor miRNAs obtained from miRBase v.21. The miRDeep2 algorithm was used to predict potential hairpin structures and to assign scores that represented the probability that hairpins were true miRNA precursors. The read counts for each miRNA were extracted from mapped miRNAs to report the abundance of each miRNA. To filter differentially expressed miRNAs, the reads for each miRNA were normalized to the total reads of each individual sample, standardized as the read per kilobase of transcript per million mapped reads (RPKM). MiRNAs with a median fold change of |fc| ≥ 2 were considered meaningful. An mRNA sequencing was done on the Illumina HiSeq 2000 platform, using 101-bp paired-end sequencing with a fragment size of 200-400 bp. The TruSeq RNA kit was used for preparing the library. Samples with the desired minimum of 5 GB mapped and properly paired read pairs from any standard mapper were chosen, without filtering for mapping quality. The mRNAs with expression profiles that differed between the samples were normalized as fragments per kilobase of transcript per million mapped reads. An mRNA with was fc ≤ –2 was considered downregulated whereas that with fc ≥ 2 was considered upregulated.
3. Primary glioblastoma tissues
Human glioblastoma tissue specimen was selected from the Department of Pathology, Ewha Womans University Mokdong Hospital, over the past 3 years (2012-2014). Hematoxylin-eosin–stained slides were reviewed and all cases showed variable degree of geographic necrosis. This study protocol was approved by the Institutional Review Board (IRB) of the Ewha Womans University Mokdong hospital (IRB protocol number: EUMC 2016-01-005). Informed consent had been obtained from each patient prior to surgery and was waived in this study by IRB due to the retrospective nature. Small RNA was isolated from formalin-fixed, paraffin-embedded (FFPE)-GBM tissues including the near-necrosis tumor area and non-necrosis tumor area using an exosomal miRNA purification kit (Genolution, Seoul, Korea) according to the manufacturer’s protocol.
4. Reverse transcription–polymerase chain reaction
cDNA was synthesized from 1 μg of total RNA with reverse transcription (MLV-RT, Enzynomics, Daejeon, Korea) and polymerase chain reaction (PCR) was performed 3 μL cDNA with a thermal cycler (Bio-Rad Laboratories, Hercules, CA). The mRNA levels were determined using G-Taq DNA polymerase, 10× Taq buffer and 2.5 mM dNTP (Labo-Pass, COSMO Genetech, Seoul, Korea). Primer sequences are listed in Table 1. Each PCR samples were visualized on 1% agarose gels stained with GelRed (Biotium Inc., Hyward, CA).
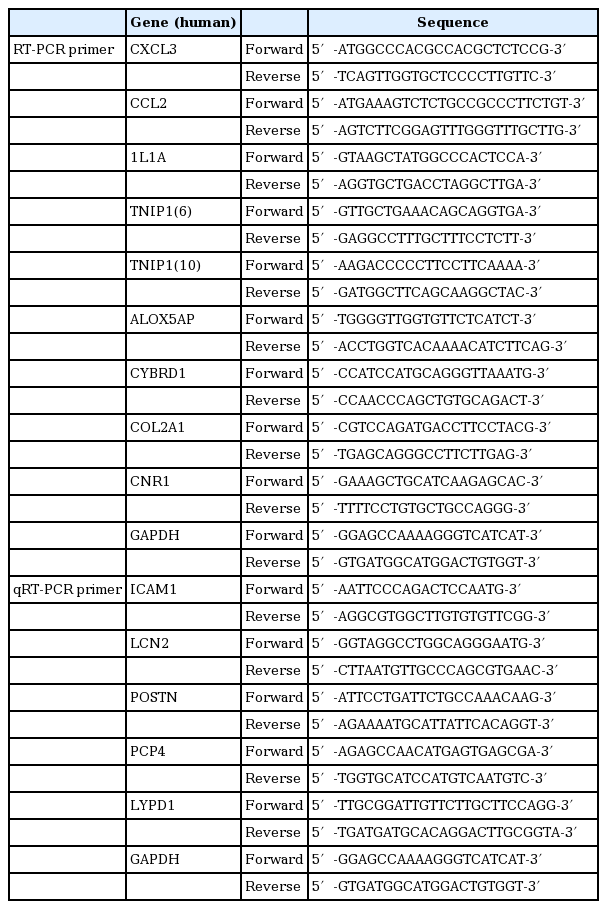
List of primer sequences used for RT-PCR and qRT-PCR analysis
5. Quantitative reverse transcription–polymerase chain reaction
Quantitative reverse transcription–polymerase chain reaction (qRT-PCR) was performed on an ABI Step One Plus Real-Time PCR machine (Applied Biosystems, Foster City, CA) using a Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer’s protocol. Primer sequences are listed in Table 1. Quantitation approach was performed by termed the comparative Ct method as previously described [21]. The Ct values of both the control and the samples of interest are normalized to an appropriate GAPDH gene. Relative expression levels were calculated as 2−ΔΔCt method, where ΔΔCt=target gene sample ΔCt–target gene control ΔCt.
6. miRNA-mRNA integrative genomic analysis
For identification of the candidate miRNA-mRNA network, three separate steps were carried out. First, miRNAs and mRNAs with differential expression were extracted though the differentially expressed gene and differentially expressed miRNA list, with significant cut-offs miRNA |fc| ≥ 2 and mRNA |fc| ≥ 2 in the miRNA and mRNA data set. Second, the putative miRNA-mRNA regulatory relationship was inferred through target information, using miRDB (v.5.0; http://mirdb.org/miRDB/). Finally, the negative correlation between the miRNA and mRNA expression profiles was confirmed.
7. Functional analysis
Based on the miRNA-mRNA integrative genomic analysis, we identified biological themes for each negatively correlated miRNA-mRNA pair using Gene Ontology (GO) (http://geneontology.org/). GO provides annotation terms that are arranged hierarchically in three namespaces that describe different aspects of a protein’s function: molecular function, biological process, and cellular component (http://geneontology.org/page/ontologydocumentation) [22]. Pathway analysis of negative miRNA-mRNA correlations was mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.kegg.jp/kegg/pathway.html). KEGG includes a pathway database with computerized knowledge on molecular interaction networks, such as pathways and complexes. The p-value was calculated for each enriched pathway [23].
8. Statistical analysis
The mean±standard error of the mean from two independent experiments is shown in all bar graphs. Statistical significance was analyzed with Student’s t test, using the program R Statistical Language v.3.1.2 (Institute for Statistics and Mathematics, Vienna, Austria). Statistical significance of the data was set at a value of p < 0.05.
Results
1. Differentially expressed miRNAs between necrotic cell-treated and untreated CRT-MG cells
To test whether necrotic tissues affect the miRNA-mRNA regulatory network in GBM, CRT-MG cells were either untreated or treated with necrotic cells and mature miRNAs and mRNA were quantitatively analyzed using NGS. Necrotic CRT-MG cells were prepared by freezing/thawing methods as described previously and confirmed by flow cytometry revealed that more than 90% of prepared cells were propidium iodide–positive [5]. The expression of mature miRNAs using small RNA sequencing. Of the 2,588 mature miRNAs detected in the analysis, 1,908 with at least one zero raw signal (RPKM) were excluded and 680 mature miRNAs for further analysis. Compared with untreated cells, the treated CRT-MG cells showed dysregulation of 106 miRNAs that had significantly different expression levels with 2-fold change as a cut off (Fig. 1A). Of these 106 miRNAs, 55 were upregulated (52%) and 51 were downregulated (48%) in the treated cells (Fig. 1B). The top five mature miRNAs are marked in the scatter plot; miR-4792, miR-146a-5p, and miR-155-5p were upregulated, and miR-let-7e-5p and miR-let-7c-5p were downregulated (Fig. 1C). The top 20 of the 55 upregulated miRNAs and the top 20 of the 51 downregulated miRNAs, as well as miRNAs matched to previous studies that have been previously reported in GBM are summarized in Table 2 [24-27]. To explore whether miRNA expression is altered in primary GBM tissues, we performed qRT-PCR for four miRNAs using FFPE glioblastoma patient samples. In miRNA real-time PCR analysis using primary GBM tissues, we observed similar finding that followed the sequencing trend in CRT-MG cells, showing increased miR-3656 and decreased miR-874-5p expression in perinecrotic region compared to non-necrosis tumor tissues (Fig. 1D).
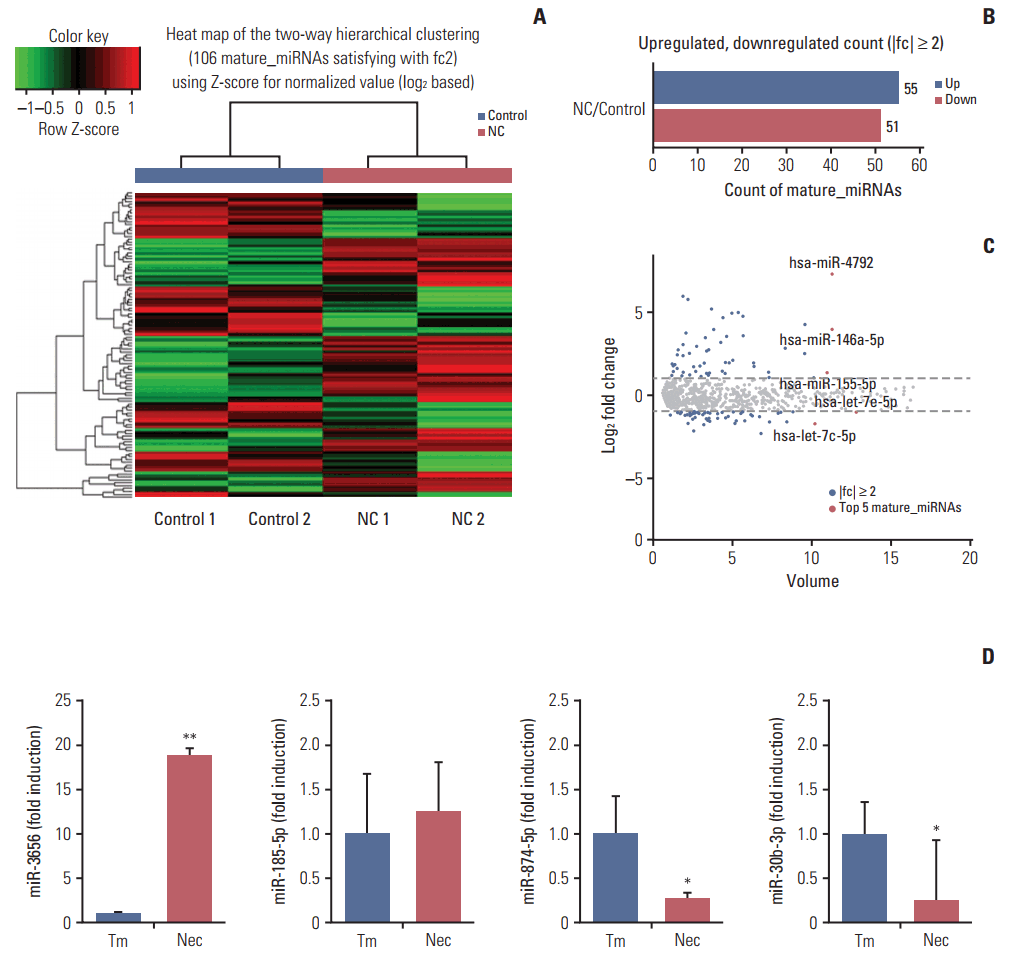
Graphical representation of the 106 miRNAs differentially expressed between necrotic cell-treated and untreated CRT-MG cells. (A) Clustering of the 106 most differentially upregulated and downregulated miRNAs for classification between the necrotic cell–treated (NC) and untreated CRT-MG cells (control). Complete linkage hierarchical clustering was performed with the Euclidian distance measure. The NC and control cells clustered separately. The colors in the heat map represent the normalized expression values, with lower expression values being colored in shades of green and higher expression values in shades of red. (B) Counts of mature miRNAs upregulated or downregulated in NC cells. (C) Scatter plot showing the normalized miRNA volume of all conditions analyzed (y-axis) and changes in miRNAs expression between the NC and control cells. The top five most significantly dysregulated miRNAs are marked. (D) Quantitative real-time polymerase chain reaction analysis was performed glioblastoma multiforme tissues from the near-necrosis tumor area (Nec) and from non-necrosis tumor area (Tm). Data are presented as fold induction compared with non-necrosis tumor area. *p < 0.05, **p < 0.01 vs. Tm.
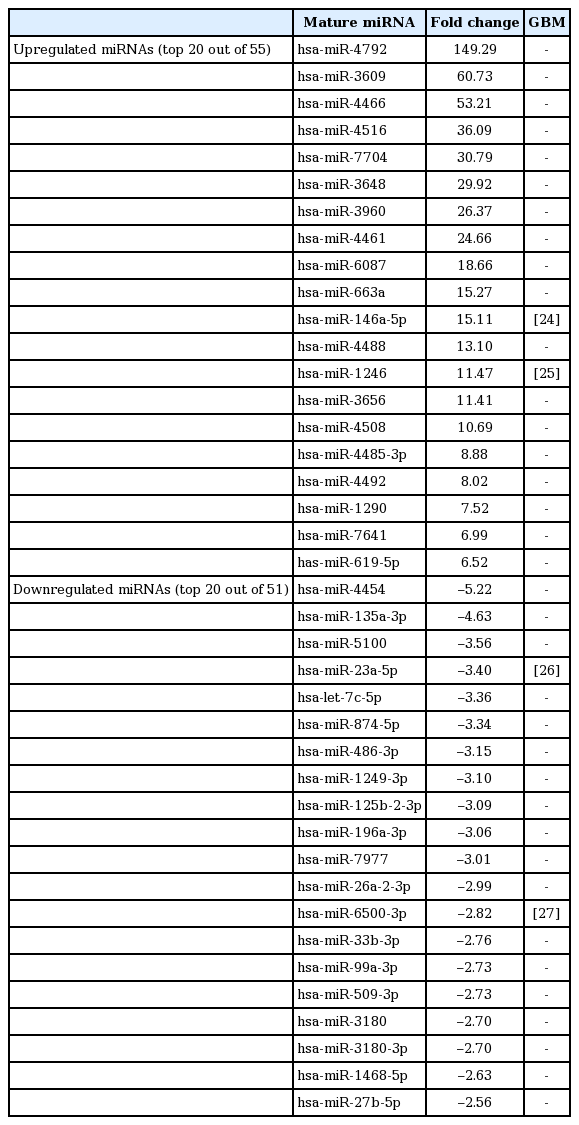
The top 20 most differentially upregulated and downregulated miRNAs in necrotic cell.treated CRT-MG cells
2. Differentially expressed mRNAs between necrotic cell-treated and untreated CRT-MG cells
Next, we investigated the expression patterns of mRNAs using transcriptome resequencing. Of the 25,906 genes detected, 7,475 with at least one zero raw signal (fragments per kilobase of transcript per million mapped reads [FPKM]) were excluded, leaving 18,431 genes to be further analyzed. These mRNA expression profiles revealed significant dysregulation of 887 mRNAs in the treated CRT-MG cells (Fig. 2A). We further found differences in the classification of mRNAs in the treated cells relative to the untreated cells. As shown in Fig. 2B, 508 mRNAs were upregulated (57%), whereas 379 mRNAs were downregulated (43%), after incubation with necrotic cells. The top five genes are indicated in the scatter plot; IL32, MT2A, and FTH1 were upregulated, and KRT18 and KRT8 were downregulated in the treated cells (Fig. 2C). The top 20 most differentially upregulated and downregulated mRNAs in the treated cells, and the mRNAs matched to existing reports about GBM, are shown in Table 3 [28-50]. We also utilized reverse transcription–polymerase chain reaction (RT-PCR) and qRT-PCR for direct validation of differential expression of mRNA in necrotic cell treated and untreated CRT-MG cells (Fig. 2D and E).
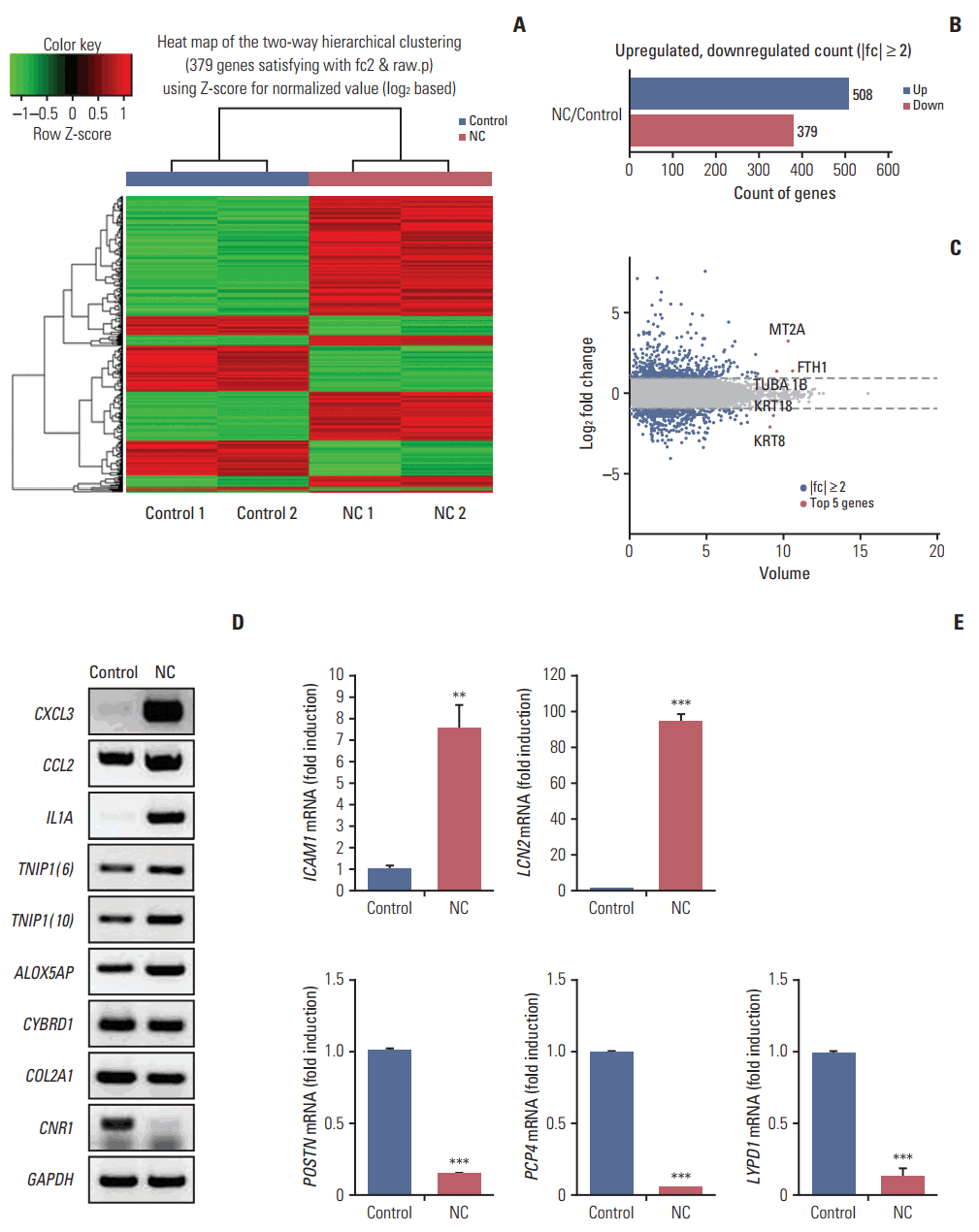
Graphical representation of the 887 mRNAs differentially expressed between necrotic cell–treated (NC) and untreated CRT-MG cells. (A) Clustering of the 887 most differentially upregulated and downregulated mRNAs for classification between the NC and untreated CRT-MG cells (control). Complete linkage hierarchical clustering was performed with the Euclidian distance measure. The NC and control cells clustered separately. The colors in the heatmap represent the normalized expression values, with lower expression values being colored in shades of green and higher expression values in shades of red. (B) Counts of mRNAs upregulated or downregulated in NC cells. (C) Scatter plot showing the normalized mRNA volume of all conditions analyzed (y-axis) and changes in mRNA expression between the NC and control cells. The top five most significantly dysregulated genes are marked. (D, E) Validation of mRNA analyzed reverse transcription–polymerase chain reaction (RT-PCR) and quantitative reverse transcription–polymerase chain reaction (qRT-PCR). (D) The expression level of six upregulated mRNA (CXCL3, CCL2, IL1A, TNIP1 [transcript variant 6 and 10], and ALOX5AP) and three downregulated mRNA (CYBRD1, COL2A1, and CNR1) were validated by RT-PCR. GAPDH gene was evaluated as external control. (E) Two upregulated mRNA (ICAM1 and LCN2) and three downregulated mRNA (POSTN, PCP4, and LYPD1) were determined by qRT-PCR. Data are presented as fold induction compared with untreated necrotic cells. **p < 0.01, ***p < 0.001 vs. control.
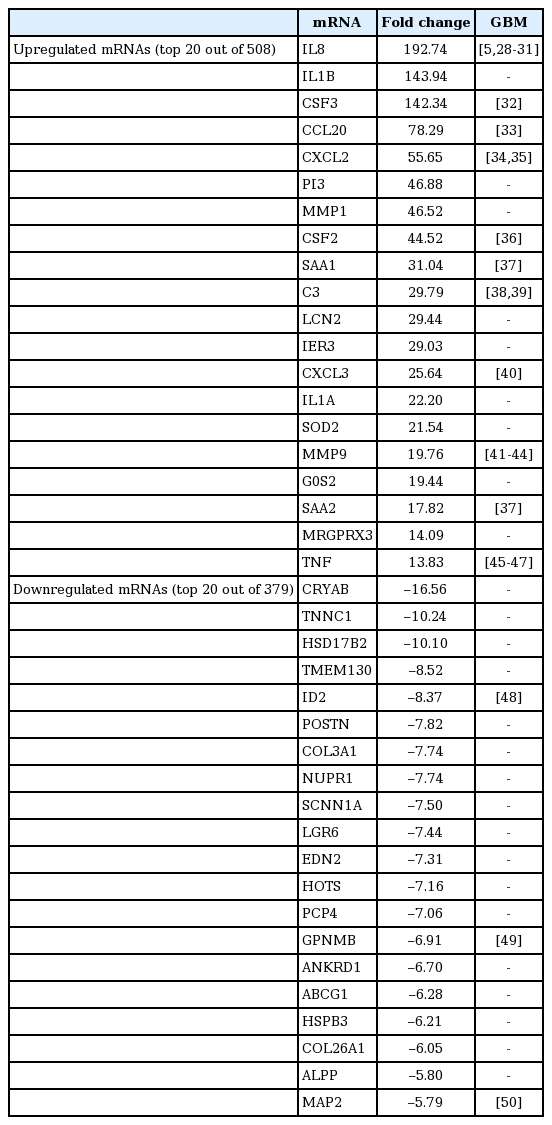
The top 20 most differentially upregulated and downregulated mRNAs in necrotic cell.treated CRT-MG cells
3. Integrative genomic analysis of negative miRNA-mRNA correlations in necrotic cell-treated CRT-MG cells
To understand the potential functions of the significant differentially expressed miRNAs, and to further explore miRNA-mRNA regulatory information in the treated cells, we performed an analysis of the gene expression data to identify genes that were inversely co-expressed with an miRNA, as well as analysis of the target prediction that was used to identify genes containing binding sites of an miRNA. The 106 mature miRNAs and 887 validated mRNAs that were differentially expressed in the treated CRT-MG cells were analyzed by searching all the miRNAs in the miRDB v5.0 database (mirBase rel.21). Consequently, 88 miRNAs were found to have at least one negatively regulated miRNA-mRNA pair for significant mRNAs, and 11 miRNAs a negative correlation with p < 0.05 by the hypergeometric test (Table 4). The upregulated miRNA-downregulated mRNA and downregulated miRNA-upregulated mRNA pairs in necrotic cell-treated CRT-MG cells with p < 0.05 by the hypergeometric test are shown in Fig. 3. Detailed information about each miRNA-mRNA integrative genomic analysis is shown in Table 5.

Summary of the miRNA-mRNA integrative genomic analysis
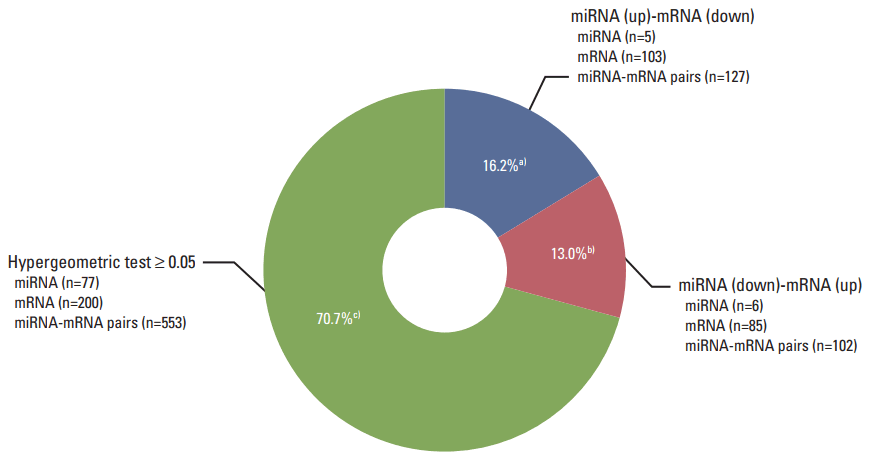
Pie chart representing the distribution of negatively correlated miRNA-mRNA pairs. Pie chart depicting the distribution of negatively correlated miRNA-mRNA pairs within the three color-coded groups. Eighty-eight miRNAs were found to have at least one negatively regulated miRNA-mRNA pair for significant mRNAs, and 11 miRNAs a negative correlation with p < 0.05 by the hypergeometric test. a)Upregulated miRNAs–downregulated mRNAs in necrotic cell–treated CRT-MG with p < 0.05 by the hypergeometric test, b)Downregulated miRNAs–upregulated mRNAs in necrotic cell–treated CRT-MG with p < 0.05 by the hypergeometric test, c)Negatively regulated miRNA-mRNA pairs with p ≥ 0.05 by the hypergeometric test.
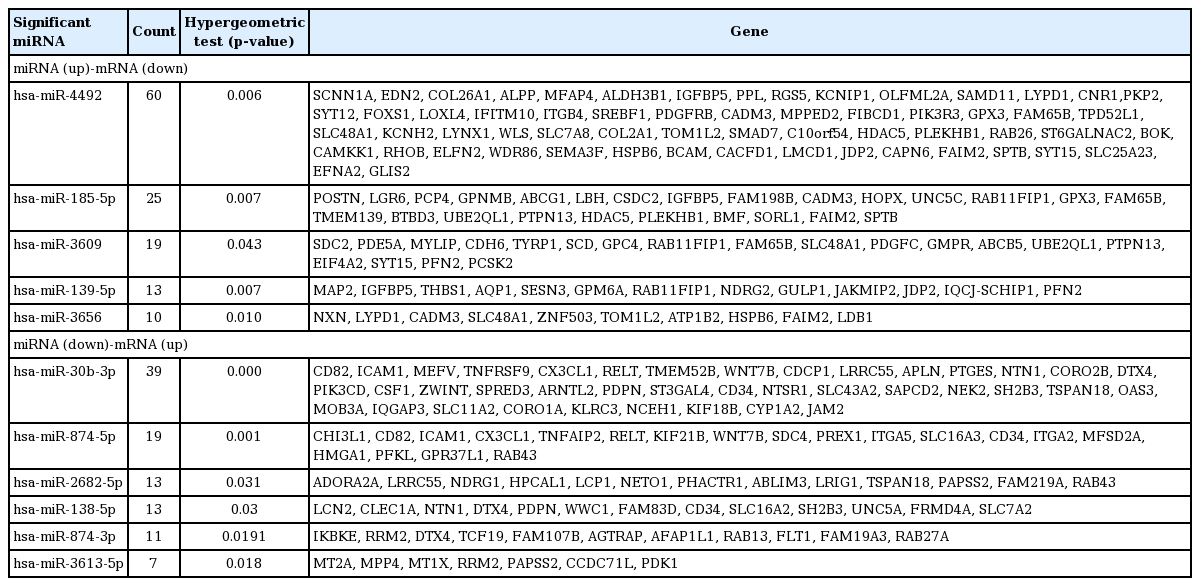
Integrative genomic analysis of 11 miRNA target genes
4. Function annotation and pathways affected by relevant negative miRNA-mRNA correlations in necrotic cell-treated CRT-MG cells
In order to study the regulatory effects of the significant negatively correlated miRNA-mRNA pairs, and to further explore the function of the predicted target genes, 188 genes that were significantly (p < 0.05) upregulated and downregulated in the treated CRT-MG cells were selected for mapping into the DAVID database and subjected to functional enrichment analysis. According to the GO enrichment analysis, these mRNAs were involved in the regulation of biologica processes, cellular components, and molecular functions (Fig. 4). The pie graph shows that 151 mRNAs were related to single-organism cellular processes in the “biological process” term, and 168 mRNAs were included in cell parts in the “cellular component” classification. Moreover, the “molecular function” category of GO terms showed that 152 mRNAs mainly played a role in protein binding, ion binding, and catalytic activity. Pathways modulated by relevant gene targets of the differentially expressed miRNAs of negatively correlated miRNA-mRNA pairs were determined using KEGG pathways. All the significant target pathways for the negatively correlated miRNA-mRNA pairs are presented in Fig. 5. Upregulation of negative miRNA-mRNA correlation was related to focal adhesion, Rap1 signaling, regulation of actin cytoskeleton, and AMP-activated protein kinase signaling pathways, whereas downregulation of negative miRNA-mRNA correlation was shown to be related to proteoglycans in cancer, central carbon metabolism in cancer, cell adhesion molecules, and cytokine-cytokine receptor interaction (Table 6).
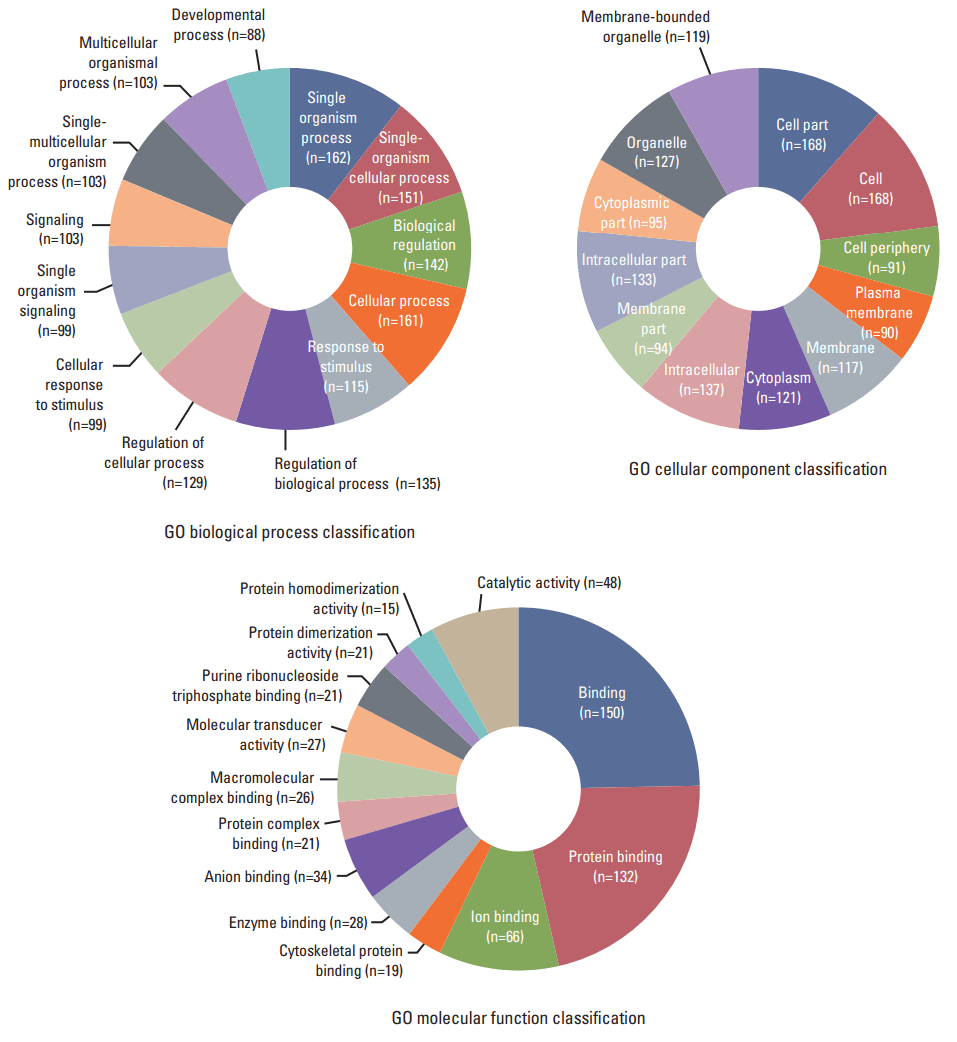
Gene Ontology (GO) enrichment analysis for negatively correlated miRNA-mRNA. The 188 genes that were upregulated and downregulated in necrotic cell–treated CRT-MG cells, with p < 0.05 by the hypergeometric test, were subjected to GO functional enrichment analysis. To study the regulatory effects of the significant negatively correlated miRNA-mRNA pairs, and to further explore the function of the predicted target genes. These mRNAs were included in the biological process, cell component, and molecular function classifications.
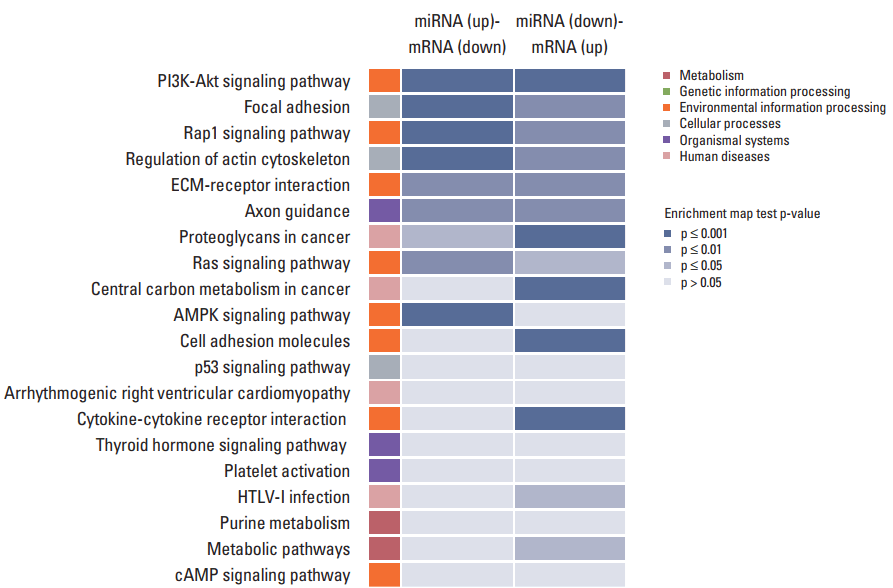
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for negatively correlated miRNA-mRNA pairs. The 188 genes that were upregulated and downregulated in necrotic cell–treated CRT-MG cells, with p < 0.05 by the hypergeometric test, were subjected to KEGG database analysis. These mRNAs were related to various biological pathways of metabolism (red), genetic information (green), environmental information (orange), cellular processes (gray), organismal system (purple), and human diseases (pink). The colors in the enrichment map represent the significant p-values of the geneset enrichment, with high significant values (p ≤ 0.001) being colored in blue shade. PI3K, phosphoinositide 3-kinase; ECM, extracellular matrix; AMPK, AMP-activated protein kinase.

KEGG pathway analysis of the genes obtained from negatively correlated miRNA-mRNA pairs
Discussion
In this study, using the human astroglioma cell line CRT-MG, we investigated the effect of necrosis on the miRNA and mRNA expression profiles and further examined the miRNA-mRNA correlations. When CRT-MG cells were exposed to necrotic CRT-MG cells, we found that 55 miRNAs were upregulated and 51 were downregulated at a significantly different expression level, with a 2-fold change as a cut off. Among them, 11 miRNAs that had a negative miRNA-mRNA correlation value of p < 0.05 by the hypergeometric test were screened, and target mRNAs were analyzed by GO enrichment analysis. Using the KEGG database, we also found that several pathways that were activated by the necrotic cell treatment were modulated by relevant gene targets of differentially expressed miRNAs with negative miRNA-mRNA correlation.
Exposure to necrotic cells induced the dysregulation of several miRNAs and the differential expression of mRNAs in the CRT-MG cells. The examination revealed that miR-4492 was increased and showed negative regulation with 60 downregulated target mRNAs through the necrotic cell treatment (Table 5). Since the effect of miR-4492 on brain cancer is poorly understood at present, our results suggest that miR-4492 might be a novel prognostic marker of GBM that is activated by necrosis. The increased miR-4492 expression negatively regulated COL2A1 and CNR1 mRNAs, which are related to focal adhesion and Rap1 signaling. Rap1 is a small GTPase that regulates cell adhesion, cell-cell junction formation and cell polarity thought mitogen-activated protein kinase. Cell adhesion signaling is known to modulate cell proliferation, survival, and migration in several cancers, suggesting that this signaling is important for the development of new therapeutic targets [51-53]. We confirmed by RT-PCR that the expression levels of the COL2A1 and CNR1 genes were decreased by the treatment of necrotic cells for validation of mRNA sequencing data (Fig. 2D).
Previous studies have provided evidence of multiple relationships between miRNAs and their target genes in cancer [14,54]. It appears that the dysregulation of miRNA profiles could serve as an effective diagnostic and prognostic biomarker. In our systems, we found the significant dysregulation of the expression of 11 miRNAs in necrotic cell-treated CRT-MG cells compared with untreated cells. Similar to previous reports about GBM, we found that miR-146a-5p and miR-1246 were upregulated in the treated CRT-MG cells [24,25]. While the expression level and function of miRNAs in glioblastoma have been extensively studied by several groups, the effects of necrosis/necrotic cells on miRNA and mRNA expression and necrosis-related cellular pathways have not been investigated. Further studies are needed to identify cellular pathway within necrosis-related GBM. We were also able to identify the dysregulation of novel miRNAs in the treated astroglioma cells. In particular, the miR-4792 level was increased by 149-fold, and miR-3609 by 60-fold, in the presence of necrotic cells, suggesting that these miRNAs could be potential candidates for the detection of necrosis.
Using NGS, we confirmed that not only miRNA expression but also mRNA expression was significantly modulated by necrotic cells in CRT-MG cells, as controlled by a variety of gene regulatory factors, including transcription factors and miRNAs. Incubation with necrotic cells induced the dysregulation of 887 mRNAs (Fig. 2). Among them, 188 mRNAs had a negative correlation with 11 miRNAs. For example, the dysregulation of PFN2, WNT7B, PFKL, PDK1, and LCN2 mRNAs was negatively correlated with miR-3609, miR-30b-3p, miR-874-5p, miR-3613-5p, and miR-138-5p. On the other hand, the expression levels of IL-8, IL-1β, CSF3, CCL20, and CXCL2 mRNAs, which were included within the 887 mRNAs deregulated by necrotic cell treatment, were remarkably increased (Table 3); however, we did not find significant negative correlations between these mRNAs and their corresponding miRNAs. Recently, we reported that when GBM cells are exposed to and stimulated by necrotic cells, activation of the transcription factors nuclear factor κB and activator protein 1 and their binding to the interleukin 8 (IL8) promoter were induced, leading to enhanced IL8 mRNA expression and protein secretion in GBM cells [5]. These results imply that necrotic cells had a regulatory effect on mRNA expression by both miRNA-mediated gene regulatory and transcription factor-mediated events.
It has been reported that negatively regulated miRNA-mRNA pairs have a significant effect on the prognosis of cancer [55,56]. In this study, we used KEGG to identify pathways that were related to negative miRNA-mRNA correlation. Consequently, we identified key miRNAs such as miR-874-5p and miR-30b-3p that were implicated in proteoglycan, central carbon metabolism, and cytokine-cytokine receptor interaction pathways, based on their target genes, such as PIK3CD and CX3CL1 (Table 6). PIK3CD is a phosphoinositide 3 kinase class I isoform that is found in a number of immune cells. In metastatic colorectal carcinoma and GBM, PIK3CD is upregulated via the downregulation of miRNA, which regulates the Akt pathway [57,58]. Moreover, the chemokine ligand CX3CL1 (also known as fractalkine) is expressed mainly in monocytes during inflammatory conditions [59]. Previous studies have demonstrated that CX3CL1 acts as a positive modulator of breast cancer, and also functions as an inflammatory mediator for communication between neurons, glia, and microglia during inflammation and neuro degeneration [60-62]. We identified the negatively regulated miR-874-5p and miR-30b-3p/PIK3CD and CX3CL1 pairs, which were implicated in proteoglycan and cytokine-cytokine receptor interaction signaling pathways, suggesting them to be novel prognostic markers in GBM that are exposed and activated by necrosis. Further study will be required to address the clinical relevance of these miRNAs and mRNAs and their putative role on necrosis-related molecular pathways for the development of novel diagnostic and prognostic biomarkers.
In conclusion, our study demonstrate that when GBM cells were exposed to and stimulated by necrotic cells, the expression profiles of miRNA and mRNA were significantly dysregulated. This is the first study to address the effect of necrotic cells on miRNA-mRNA expression and association in GBM cells. The data implicated several miRNAs including miR-4492 might be a novel prognostic marker of GBM that is activated by necrosis.
Notes
Conflict of interest relevant to this article was not reported.
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) Grant 2010-0027945 and by NRF-2016R1A2B4016376.