Outcomes of Treatment for Malignant Peripheral Nerve Sheath Tumors: Different Clinical Features Associated with Neurofibromatosis Type 1
Article information
Abstract
Purpose
Malignant peripheral nerve sheath tumors (MPNSTs) are a rare subtype of sarcoma that occur spontaneously or in association with neurofibromatosis type 1 (NF-1). This study aimed to clinically differentiate these types of MPNSTs.
Materials and Methods
The study reviewed 95 patients diagnosed with and treated for MPNST at Yonsei University Health System, Seoul, Korea over a 27-year period. The clinical characteristics, prognostic factors, and treatment outcomes of sporadic MPNST (sMPNST) and NF-1 associated MPNST (NF-MPNST) cases were compared.
Results
Patients with NF-MPNST had a significantly lower median age (32 years vs. 45 years for sMPNST, p=0.012), significantly larger median tumor size (8.2 cm vs. 5.0 cm for sMPNST, p < 0.001), and significantly larger numbers of imaging studies and surgeries (p=0.004 and p < 0.001, respectively). The 10-year overall survival (OS) rate of the patients with MPNST was 52±6%. Among the patients with localized MPNST, patients with NF-MPNST had a significantly lower 10-year OS rate (45±11% vs. 60±8% for sMPNST, p=0.046). Univariate analysis revealed the resection margin, pathology grade, and metastasis to be significant factors affecting the OS (p=0.001, p=0.020, and p < 0.001, respectively). Multivariate analysis of the patients with localized MPNST identified R2 resection and G1 as significant prognostic factors for OS.
Conclusion
NF-MPNST has different clinical features from sMPNST and requires more careful management. Further study will be needed to develop specific management plans for NF-MPNST.
Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a type of soft tissue sarcoma that originates from the peripheral nerve sheath. These tumors account for 5%-10% of all soft tissue tumors, but their occurrence is rare in the general population with an estimated annual incidence of 1/1,000,000 individuals [1-3]. MPNST is frequently associated with neurofibromatosis, and has an incidence of 2%-5% among patients with neurofibromatosis type 1 (NF-1); this incidence is very high relative to that of the general population [2]. MPNST is considered aggressive and is associated with a low survival rate (34%-52%) [4]. The prognosis of MPNST is associated with the tumor size and location, resection margin, adjuvant chemotherapy, distant metastasis, stage, and site [5-7].
NF-1 is an autosomal hereditary syndrome characterized by symptoms and signs, such as neurofibromas, café-au-lait spots, osseous lesions, optic pathway glioma, axillary or inguinal freckling, and Lisch nodules, with a global incidence of 1/2,500-1/3,000 individuals [8,9]. Throughout their lives, patients with NF-1 frequently develop benign and malignant tumors consequent to the loss of the tumor suppressive activity of NF1 [8]. The descendants of patients with NF-1 are born with a systemic heterozygous NF1 mutation; however, a mutation of the second normal copy is required for the formation of NF1-related tumors [10]. Patients with NF-1 carry an approximately 2.5- to 4-fold higher risk of malignancy relative to the general population, as well as the risk of optic glioma and soft tissue sarcoma, which have been reported to occur in 15%-20 % and 4%-25% of patients, respectively [8].
The prognostic role of a patient’s NF-1 status with regard to the MPNST treatment has been studied because the genetic backgrounds underlying sporadic MPNST (sMPNST) and MPNST arising in patients with NF-1 (NF-1 associated MPNST, NF-MPNST) differ, and the clinical manifestations of NF-1 frequently include multiple benign and malignant tumors [6,11,12]. On the other hand, the differences in survival associated with these different subtypes is controversial [6,11-16]. Although many researchers have reported the characteristic features of NF-MPNSTs, there are no standard management guidelines for this subtype. The present study evaluated the clinical differences between the two subtypes of MPNST to further the development of specific diagnostic and treatment strategies for NF-MPNST.
Materials and Methods
1. Patient population
A total of 95 patients diagnosed with and treated for MPNST at the Yonsei Cancer Center, Yonsei University Health System, Seoul, Korea from 1988 to 2015 were enrolled in this study. The clinical data were collected via retrospective chart review. A clinical diagnosis of NF-1 was confirmed by the presence of one of the following criteria: confirmed NF1 mutation and associated symptoms, or ≥ 2 clinical manifestations that met the National Institutes of Health (NIH) consensus criteria [3,9]. The direct polymerase chain reaction sequencing method of the patient DNA isolated from a blood sample was used for a genetic NF1 test. Among the 33 patients, 32 met the clinical diagnosis criteria fully. Among them, an additional genetic test was performed for sixpatients. For only one patient who did not meet the clinical diagnostic criteria, the genetic test was used to confirm NF-1. NF-MPNST was defined as MPNST that occurred in a patient diagnosed with NF-1, and sMPNST was defined as MPNST arising in a patient without NF-1. This study was approved by the Institutional Review Board of Severance Hospital, Yonsei University Health System (No. 4-2016-0401).
2. Treatment
Surgery was generally performed with curative intent. Palliative surgery was performed to relieve the symptoms caused by the tumor. A biopsy alone was performed in two cases involving unresectable lesions. Radiotherapy was generally administered to patients with positive tumor margins after surgery or to reduce symptoms, such as pain or other neurologic signs. Chemotherapy was generally administered for metastatic MPNST or recurrent disease. First-line adjuvant treatments for localized MPNST were evaluated as treatment patterns.
3. Clinical data
The pathologic diagnoses of MPNST were initially made but were reviewed by the institutional pathologist at the time of the study in accordance with the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) pathologic grading system [17]. The FNCLCC system considered the cellularity, nuclear pleomorphism, anaplasia, mitotic rate, necrosis, and microvascular proliferation [17]. For 15 of the 95 patients, the grades could not be defined either because these patients had been referred from other institutions (n=10) or tissues were unavailable because the diagnoses had been made at least 20 years earlier (n=5).
Information on the chemotherapeutic regimens was collected from the medical records. Data regarding the tumor size and depth, lymph node involvement, and distant metastases were collected from imaging studies, which included computed tomography, magnetic resonance imaging, ultrasonography, and positron emission tomography. This study evaluated the total number of imaging studies or operations including biopsies, which are defined as the sum of the number of imaging studies or operations performed between the time of diagnosis and the last follow-up. Disease progression was defined as tumor growth after surgical intervention, radiotherapy, or chemotherapy, based on imaging studies. Staging information according to the seventh American Joint Committee on Cancer (AJCC) staging system was obtained from the surgical records [18]. The margin status was defined as follows: R0, microscopically negative margins (tumor-free margin > 2 mm); R1, macroscopically negative margins with microscopically positive margins; or R2, macroscopically positive margins.
4. Statistics
The overall survival (OS) was calculated as the interval from the date of diagnosis to the date of death from any cause or the last follow-up. OS was calculated using the number of deaths prior to April 30, 2016. A chi-square test or Fisher exact test was used to analyze the categorical variables, and a Student's t test or Mann-Whitney test and an analysis of variance (ANOVA) or the Kruskal-Wallis test were used to analyze the continuous variables. The Kaplan-Meier method was used for survival analyses, and the results were compared using a log-rank test. A Cox proportional hazard regression model was used for multivariate data analysis and included the factors identified as significant in univariate analysis. Age was also included because this factor was considered clinically meaningful with respect to the treatment tolerability and hereditary cancer syndrome. SPSS ver. 20 for Windows (IBM Corp., Armonk, NY) was used for the statistical analysis of each outcome measure.
Results
1. Clinical characteristics
The overall median age was 40.4 years (interquartile range, 28.3 to 54.0 years; range, 8.3 to 80.4 years) (Table 1). In particular, the 33 patients (35%) with NF-MPNST had a significantly lower median age (31.9 years vs. 45.3 years for sMPNST, p=0.012). Forty-five patients (47%) in the overall group were female, and no significant sex-related difference was observed between the patients with sMPNST and NF-MPNST.

Demographic characteristics of patients with malignant peripheral nerve sheath tumors
Overall, the tumors presented most frequently in the extremities (n=45, 47%), followed by the trunk (n=34, 35%) and head and neck (n=16, 17%). Among patients with NF-MPNST, the trunk was the most common tumor site (n=15, 46%), whereas the extremities were the most common sites among patients with sMPNST (n=31, 50%). On the other hand, these inter-group differences in MPNST sites were not significant (p=0.325). The median size of the primary NF-MPNSTs was greater than that of the sMPNSTs (8.2 cm vs. 5.0 cm, p < 0.001), and the proportion of large primary tumors (size > 5 cm) was significantly higher among the patients with NF-MPNST than those with sMPNST (28 [85%] vs. 26 [42%], p < 0.001).
Generally, most tumors were located deep beneath the fascia (88% overall; 89% for sMPNST vs. 88% for NF-MPNST, p=0.904). The histologic grade was similar in the two groups (p=0.302, linear by linear association). In contrast, the NF-MPNST patients were significantly more likely to have a higher tumor stage (p=0.002, linear by linear association) (Table 1). Among the cases of localized MPNST, the frequency of patients with NF-MPNST who had positive tumor margins comprised the majority of such patients and was significantly higher than the frequency among patients with sMPNST (66.7% vs. 47.6%, p=0.035). Similar frequencies of metastases were observed in both groups (6 [10%] vs. 3 [9%], p=0.926).
2. Management patterns
Overall, 93 patients (98%) underwent primary surgery. Only two patients (2%) underwent a biopsy alone. In addition, 46 patients (48%) received radiotherapy, and 27 (28.4%, 27/95) received chemotherapy; of these, 16 had sMPNST. Seven patients (28%, 7/25) received neoadjuvant chemotherapy. Anthracyclines (15%), ifosfamide (27%), cisplatin (17%), and etoposide (5%) were the most commonly used chemotherapeutic agents, and the most common regimen was anthracycline+ifosfamide (41%, 11/27), followed by ifosfamide+cisplatin+etoposide (26%, 7/27). Only two patients were treated with anthracycline+ifosfamide+cisplatin (7%, 2/27), and one patient used anthracycline+cisplatin and ifosfamide+cisplatin (S1 Table).
The first-line treatment schemes administered to patients with localized sMPNST and NF-MPNST were compared (Fig. 1). In the R0 group, 60% (18/30) of patients with sMPNST received no further therapy, whereas 56% (5/9) with NF-MPNST received adjuvant treatments (p=0.202). In the R1 group, 50% (4/8) of patients with sMPNST received no treatment, whereas 75% (6.8) of those with NF-MPNST received further adjuvant treatments (p=0.315). In the R2 group, 9% (1/11) of patients with sMPNST and 30% (3/10) with NF-MPNST received chemotherapy and radiotherapy both as the adjuvant treatment (p=0.181). When the analysis was limited to localized MPNST in total, 76 patients (49 sMPNSTs and 27 NF-MPNSTs) underwent a primary resection. Among them, 38% (n=29) received no adjuvant treatment, 54% (n=41) received chemotherapy or radiotherapy, and 8% (n=6) received both treatments. A total of 45% (22/49) of patients with sMPNST received no further treatment, whereas 74% (20/27) of those with NF-MPNST received adjuvant treatment, including chemotherapy, radiotherapy, or both (p=0.04999). In addition, the NF-1 patients underwent a higher median annual number of imaging studies (7.54 vs. 3.67 for sMPNST, p=0.004) and operations, including biopsy (2.45 vs. 0.70, p < 0.001) during the follow-up period (S2 Table).
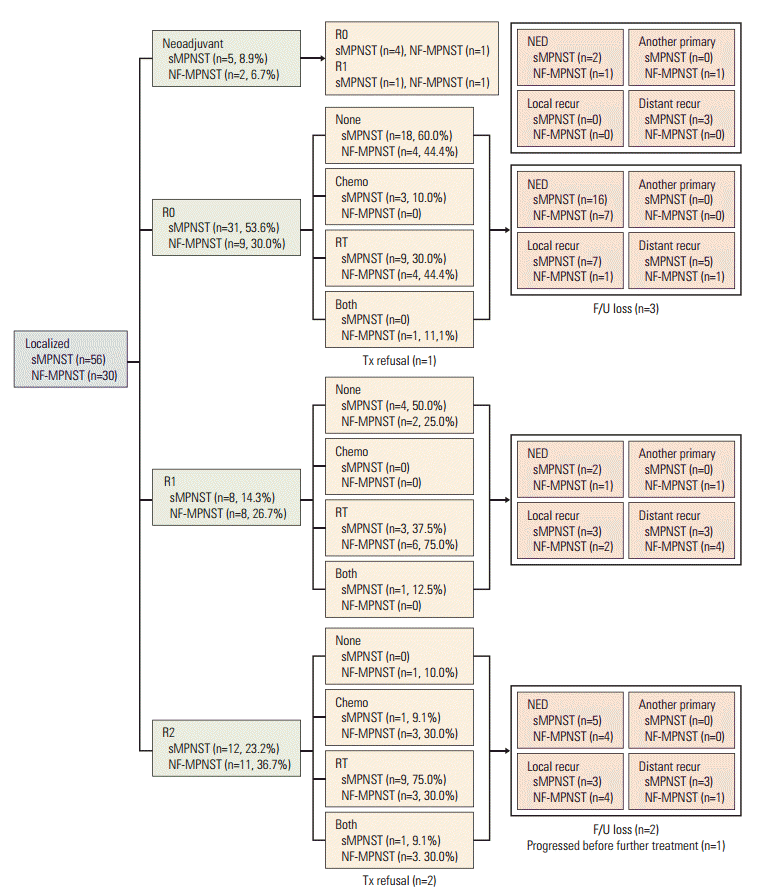
Scheme of first-line treatment modalities. sMPNST, sporadic malignant peripheral nerve sheath tumors; NF-MPNST, malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1; Chemo, chemotherapy; RT, radiotherapy; Both, chemotherapy and radiotherapy; NED, no evidence of disease; R0/R1/2, resection margin, see details in Materials and Methods section; Tx, treatment; F/U, follow-up.
3. Survival outcomes
The 10-year OS rate of the entire study cohort was 52±6% (Fig. 2). In particular, although the 10-year OS rate was lower among patients with NF-MPNST than among those with sMPNST, this difference was not significant (44±11% vs. 56±7%, p=0.084) (S3 Fig.). On the other hand, among patients with localized MPNST, those with NF-MPNST had a significantly lower 10-year survival rate (45±11% for NF-MPNST vs. 60±8% for sMPNST, p=0.046) (Fig. 3A). In contrast, patients with metastasis had similar survival outcomes, regardless of the subtype (5-year OS, 33±27% for NF-MPNST vs. 17±15% for sMPNST, p=0.875) (Fig. 3B).
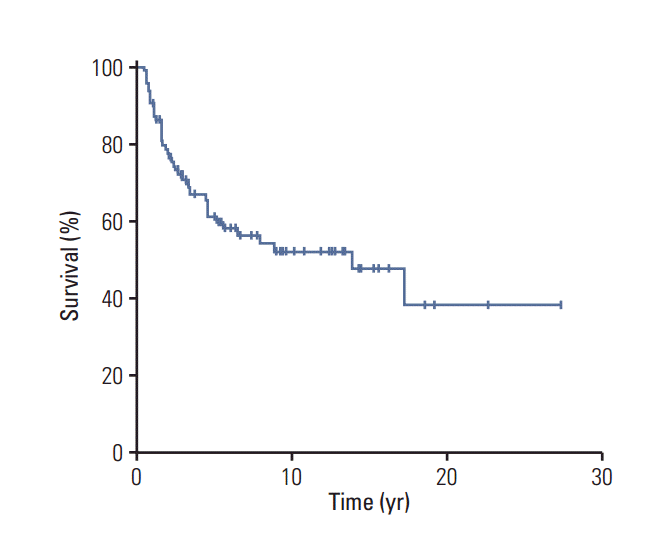
Overall survival of patients with malignant peripheral nerve sheath tumors.
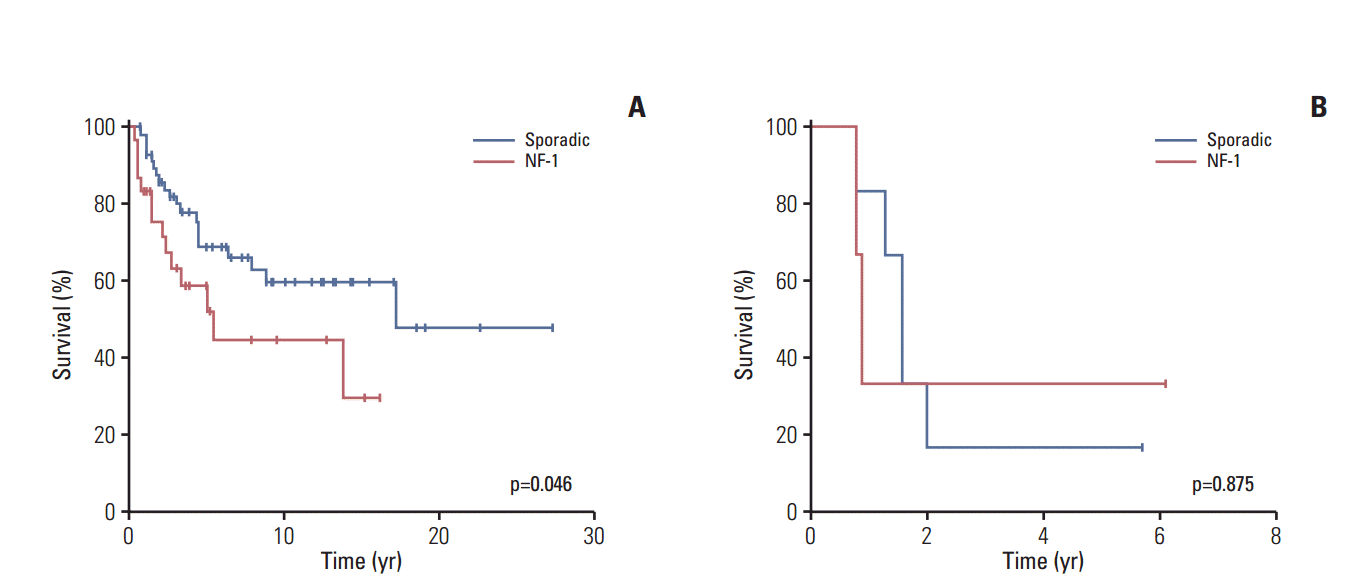
Overall survival according to neurofibromatosis type 1 (NF-1) status. (A) Overall survival associated with nonmetastatic malignant peripheral nerve sheath tumors. (B) Overall survival associated with metastatic malignant peripheral nerve sheath tumors.
Patients with R0 margins had a significantly higher 10-year OS rate than the patients with other margin statuses (65±9% for R0; 41±7% for R1 or R2; p=0.001) (Fig. 4A). The patients with pathologic G1 disease had a better 10-year OS rate than those in the other staging groups (G1 vs. G2 vs. G3 vs. N/A; 82±9% vs. 31±12% vs. 44±12% vs. 51±13%, respectively, p=0.020) (Fig. 4B). Metastasis at the time of diagnosis was associated with a poorer prognosis (5-year OS, 55±6% for localized MPNST vs. 22±14% for MPNST with metastasis, p < 0.001) (Fig. 4C). No survival difference was observed with respect to the use of a doublet vs. triplet regimen (50±14% vs. 53±15%, p=0.738) or to the tumor location and size ≥ 5 cm (p=0.264 and p=0.113, respectively; data not shown).
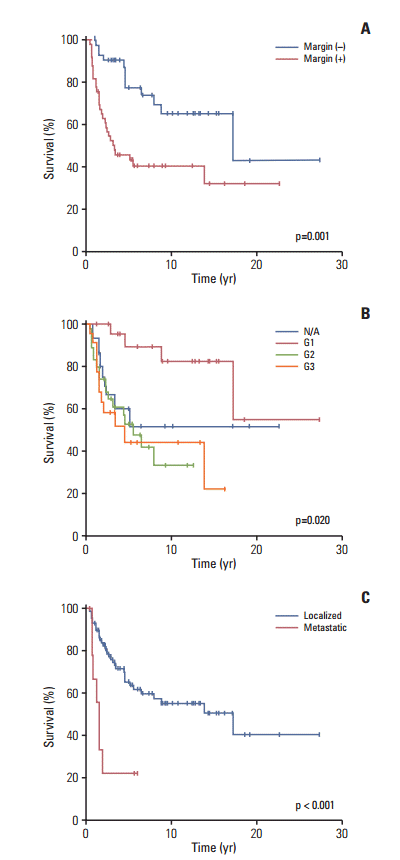
Overall survival according to the risk factors. (A) Overall survival associated with the margin status (positive vs. negative). (B) Overall survival associated with the pathologic grade (not acquired [N/A] vs. G1 vs. G2 vs. G3). (C) Overall survival associated with the metastatic status (localized vs. metastasis).
4. Multivariate analysis of clinical risk factors affecting OS in localized MPNSTs
Because NF-1 was identified as a significant prognostic factor for OS among patients with localized MPNSTs in the univariate analysis, multivariate analysis was performed to confirm the risk factors for OS. The R2 status was confirmed to be a significant risk factor for OS (hazard ratio, 2.61; 95% confidence interval [CI], 1.03 to 6.61), and histologic G1 disease was identified to be more favorable for OS compared to G3 disease (odds ratio [OR], 0.18; 95% CI, 0.05 to 0.71). On the other hand, the NF-1 status was found to be an insignificant prognostic factor for OS (OR, 1.18; 95% CI, 0.48 to 2.90). Age > 45 years and a positive resection margin were not identified as significant prognostic factors (Table 2).

Multivariate analysis of the overall survival of patients with localized malignant peripheral nerve sheath tumors
Discussion
The identified frequency of NF-MPNSTs in this study was 35%; previous studies have reported a frequencies of 20% to 70% [7,16,19-21]. NF-MPNST was found to be related to a younger age of onset, greater tumor size, and higher disease stage; these findings are in line with those of many previously reports [5,6,12,16,20,21]. Consistent with previous reports, a lower frequency of R0 resection was observed among patients with NF-MPNST compared to those with sMPNST [19,21,22]. Overall, these findings might affect the survival of patients with NF-MPNST.
In previous reports, the 10-year survival rates of MPNST ranged from 30% to 60% [6,11,12,21,23]. The observed 10-year OS rate of 52±6% was consistent with previous reports. The survival differences of patients with NF-MPNST and those with sMPNST are controversial. In the present study, the survival of patients with NF-MPNST was inferior to that of patients with sMPNST when cases of localized disease were analyzed, even though this was not confirmed in multivariate analysis. Many studies reported poorer survival among patients with NF-MPNST [5,11,20]; in contrast, others have reported no significant difference in survival between the two MPNST groups [7,10,12,16]. To the best of the authors’ knowledge, no previous report has demonstrated a better survival among patients with NF-MPNST. In other words, the survival of patients with NF-MPNST is equivalent to or poorer than that of patients with sMPNST.
Given the rarity of MPNST, few studies have been able to analyze more than 100 MPNSTs from a single institution, and existing large studies have tended to report lower survival among patients with NF-MPNST [5,6,11,23-25]. This article is similar to previous reports that also demonstrated a survival difference in univariate analysis but failed to confirm the significance of this difference in multivariate analysis [6,15]. This pattern might be due to confounding factors, such as poor prognostic factors associated specifically with NF-MPNST [5,6,15]. In particular, articles published before 2000 tended to report poorer survival outcomes among patients with NF-MPNST. Since 2000, however, the differences in survival between the two subtypes have narrowed [5,12,24,25]. The treatment protocols used prior to 2000 were different from the current treatment trends, which tended toward more intensive treatment. Over time, previous articles may represent the natural history of NF-MPNST, as well as the essential differences related to the genetic background and clinical characteristics. Despite the reduction or absence of survival differences in recent articles, the considerable heterogeneity among the study cohorts prevents a direct comparison [12,21].
As in many previous articles, metastasis was identified as a poor prognostic factor for both NF-MPNST and sMPNST [5,6,11]. Porter et al. [11] reported significant survival differences between the subtypes in an analyses of both localized and total MPNSTs, whereas Anghileri et al. [15] reported a survival difference among localized MPNSTs. Carli et al. [5] reported a survival difference between the subtypes, even in a total MPNST group (including localized and metastatic disease) and attributed this difference to the more frequent incidence of metastatic disease among patients with NF-1. In the present cohort, a significant survival difference was observed between NF-MPNST and sMPNST among localized MPNSTs, whereas only a non-specific tendency was observed in the total group. In contrast to Carli et al. [5], the frequency of metastasis was similar in patients with NF-MPN-ST and sMPNST. Hence, the survival difference between these subgroups in the total cohort might not have been evident. In addition, as metastasis might be a stronger prognostic factor than NF-1, the survival difference between the subtypes might be more evident when metastatic disease is excluded.
Patients with NF-1 tend to develop numerous benign tumors, including preexisting neurofibromas. Therefore, it is challenging for clinicians to differentiate newly developed malignancies from underlying benign tumors associated with NF-1 [8,10,12,20]. The life time risk of MPNST in NF-1 patients is high (8%-13%) [20]. This unique finding and the clinical characteristics associated with NF-MPNST affect the disease management patterns, including treatments and cancer surveillance. Regarding the treatment patterns, patients with NF-MPNST also tended to receive adjuvant treatments more frequently than patients with sMPNST. NF-MPNST is associated with a low likelihood of a R0 resection and a higher probability of a large tumor size, and clinicians tend to choose additional treatments to avoid the risk of recurrence even if a curative resections is possible [5,19,21,23]. Patients with NF-MPNST undergo both imaging studies and operations more frequently, relative to those with sMPNSTs, in an attempt by clinicians and patients to achieve an early diagnosis and resection, and overcome the challenges in distinguishing MPNSTs from benign neurofibromas [6,12,26]. All of these differences could burden both the patients with NF-1 and clinicians during the process of MPNST management [6,12,19-21]. These changes in modern management patterns might have reduced the previously mentioned survival difference between sMPNST and NF-MPNST [5,12]. Therefore, the role of these management strategies should be investigated prospectively [20].
The increased identification of genetic cancers in recent years has led to the development of agents that target genetic alterations [27,28]. NF1 acts as a tumor suppressor; hence, the genetic alteration of NF1 is among the etiologies of MPNST in patients with NF-1 [8]. On the other hand, other genetic abnormalities contribute to the malignant transformation of benign neurofibromas [29,30]. Given the previously mentioned clinical characteristics, the management of NF-MPN-ST requires the development of specific clinical prevention methods and molecular targeting agents [22,30].
The present study had some unique points, such as a relative large single-institution cohort and a focus on Asian ethnicity. On the other hand, this study was retrospective, and the gradual changes in treatment modalities and intensities over the long study period need to be acknowledged.
Conclusion
MPNSTs associated with NF-1 tend to arise at a younger age, are generally larger, and are diagnosed at a higher stage compared to sMPNST. In addition, NF-MPNST and sMPNST have different diagnostic and treatment patterns. These results highlight the need for further investigation of specific strategies that account for the differences in clinical and genetic backgrounds.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (http://www.e-crt.org).
Notes
Conflict of interest relevant to this article was not reported.