Blockade of Autophagy Aggravates Endoplasmic Reticulum Stress and Improves Paclitaxel Cytotoxicity in Human Cervical Cancer Cells
Article information
Abstract
Purpose
Autophagy is one of the ways to degrade unfolded proteins after endoplasmic reticulum (ER) stress. The purpose of this study is to determine whether a blockade of autophagy leads to aggravated endoplasmic reticulum stress, which then induces cells apoptosis in HeLa cells treated with paclitaxel.
Materials and Methods
Autophagy activation and the proapoptotic effects were characterized using monodansylcadaverine labeling and Hoechest staining, respectively. A Western blot analysis was used to detect the expression of apoptotic and autophagy-related genes. A flow cytometry was used to assess the cell apoptosis ratio.
Results
Paclitaxel exposure induced the aggregation of autophagosomes in the cytoplasms of cervical cancer HeLa cells. The expression of Beclin 1 and LC3 II were upregulated, but p62 was downregulated, which suggests that autophagy was promoted by paclitaxel. On the other hand, the expression of GRP78 obviously increased, suggesting that ER stress was induced after paclitaxel treatment. The cell proliferation assay indicated that a knockdown of Beclin 1 sensitized HeLa cells to paclitaxel. Furthermore, paclitaxel-mediated apoptotic cell death was further potentiated by the pretreatment with autophagy inhibitor chloroquine or small interfering RNA against Beclin 1. These results suggest that an induction of autophagy by paclitaxel may induce cell survival rather than cell death in HeLa cells; moreover, inhibition of autophagy led to an aggravated ER stress and an induction of downstream apoptosis.
Conclusion
Our results reveal autophagy induced by paclitaxel conferred protection of tumor cells against apoptosis, and blockade of autophagy subsequently aggravated ER stress, enhancing the apoptosis associated with paclitaxel treatment in HeLa cells.
Introduction
Paclitaxel is one of the most effective chemotherapeutic agents and is widely used in the treatment of tumors; however, its acquired resistance limits its usage. Recently, several studies have found that paclitaxel can induce autophagy. Eum and Lee [1] reported that paclitaxel induced a notable increase of autophagy in v-Ha-ras-transformed NIH 3T3 cells. In a particular study, promotion of apoptotic cell death by paclitaxel was accompanied with an induction of autophagy in A549 cells. However, the underlying mechanism is still not illustrated.
Various evidences have suggested that autophagy may play dual roles during chemotherapy. Sometimes, autophagy is a protective pathway associated with the maintenance of cellular homeostasis activated in response to nutrient deprivation and various metabolic stressful stimuli. Sometimes, autophagy is a way to induce cancer cell death [2]. Endoplasmic reticulum (ER) stress is a major regulator of autophagy. PERK, IRE1, and increased cytosolic calcium have been implicated as the mediators of ER stress induced autophagy in mammalian cells [3]. ER stress leads to evolutionarily conserved cell stress response, the unfolded protein response (UPR), which induces the expression of chaperones and proteins involved in the recovery process. Moreover, the UPR may upregulate the autophagy machinery [4,5].
ER stress can induce autophagy, and autophagy activation can degrade unfolded aggravated proteins to alleviate ER stress, thus enabling the cell to avoid ER-mediated apoptosis. Therefore, we hypothesized that the inhibition of autophagy may lead to aggravated ER stress, inducing downstream apoptosis. In this study, we tested whether autophagy suppression will exacerbate ER stress and induce ER stress related apoptosis.
Materials and Methods
1. Cell culture, drug treatment, and transfection
The human cervical cancer HeLa cell line was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) medium that contained 10% fetal bovine serum (Gibco, Germantown, MD), penicillin (10 U/mL, Sigma, St. Louis, MO), and streptomycin (0.1 mg/mL, Sigma), and were kept in a humidified atmosphere of 5% CO2 at 37°C. The cells were exposed to paclitaxel (1 μg/mL, Sigma) or chloroquine (CQ; 5 μM, Sigma) when the confluency reached 50%. Pre-designed siRNAs were purchased for Beclin-1 (Qiagen, Germantown, MD), and one negative random siRNA, exhibiting no significant sequence similarity to human, mouse, or rat gene sequence served as a negative control. The equimolar amounts of siRNAs were incubated with Lipofectamine 2000 Transfection Reagent from Invitrogen (Madison, WI) in accordance to the manufacturer’s instructions.
2. Western blot analysis
The cultured cells were washed twice with phosphate buffered saline (PBS) and lysed with a cold RIPA lysis buffer containing protease inhibitors (phenylmethylsulphonyl fluoride 1 mmol/L and leupeptin 0.1 g/L). Cell lysates were collected from culture plates using a rubber policeman, and proteins were collected by centrifugation. Protein concentrations were determined by bicinchoninic acid protein assay (Pierce, Rockford, IL). Aliquots of 40 μg of proteins were boiled in 2× loading buffer (0.1 M Tris-Cl, pH 6.8, 4% sodium dodecyl sulfate, 0.2% bromophenyl blue, and 20% glycerol) for 10 minutes, loaded into 10% Tris-HCl polyacrylamide gels, and transferred electrophoretically to Immobilon-P membrane (Millipore Corporation, Billerica, MA). The membranes were incubated with primary antibodies and appropriate horseradish peroxidase secondary antibodies. The membranes were additionally probed with an antibody against actin (Santa Cruz Biotechnology, Dallas, TX) to ensure equal loading of proteins between the samples. Detection was performed with chemiluminescent agents (Pierce).
3. Polyclonal antibodies
Anti-beclin 1 antibody (Santa Cruz Biotechnology) recognizes a single 60-kDa band on a Western blot. Anti-LC3 antibody (Santa Cruz Biotechnology) recognizes 16-kDa and 18-kDa two bands. Anti-p62 antibody (Cell Signaling, Danvers, MA), anti–glucose-regulated protein-78 (GRP78) antibody (Cell Signaling), anti-PERK antibody (Cell Signaling), anti-CHOP antibody (Cell Signaling), and anti-IRE1 antibody (Cell Signaling) recognize a single 62-kDa, 78-kDa, 140-kDa, 27-kDa, and 130-kDa band on Western blot, respectively.
4. Cell proliferation assay
HeLa cells were plated in 96-well plates at 5,000 or 6,000 cells per well, respectively. On the following day, the cells were treated with or without 1 μg/mL paclitaxel with five to six replicates for 24 hours or 48 hours. According to the experiment, the cytotoxity was assessed by incubating cells with WST-1 reagent (Beyotime, Beijing, China) for 2 hours, measuring the absorbance at 450 nm, and at 630 nm as reference, with a microplate reader (Bio-Rad, Hercules, CA). The cell survival rate was calculated as follows: (ODexperimental group– ODcontrol group)/ODcontrol group×100%.
5. Assessment of cell apoptosis by flow cytometry
Apoptosis was quantified by a combined staining of annexin V and propidium iodide (PI), using AnnexinV-FITC Apoptosis Detection Kit (Beyotime). In brief, the cells were harvested and resuspended in a 500 μL of 1× binding buffer, after treatment with paclitaxel (1 μg/mL) for 24 hours or paclitaxel treatment followed by CQ (5 μM) treatment for 48 hours. After adding 5 μL of Annexin V-FITC solution and 5 KL of PI solution, the cells were incubated for 15 minutes at room temperature in the dark. At the end of incubation, the cells were analyzed on a FACScan flow cytometry system (Becton Dickinson, San Joes, CA). Data were analyzed using a winMDIv2.8 software (Perdue University Cytometry Laboratories, West Lafayette, IN). All experiments were performed three times for each experimental condition.
6. Hoechest 33258 staining assay
Replicate cultures of 1×106 cells per well were plated in a 24-well plate. The cells were transfected with siRNA and/or treated with paclitaxel. After a change of the fresh medium 24 hours later, the cells were incubated with 5 μL of Hoechst 33258 (Beyotime) solution per well at 37°C for 10 minutes, followed by observation under a fluorescence microscope. Strong fluorescence can be observed in the nuclei of apoptotic cells, while weak fluorescence was observed in non-apoptotic cells. Quantification of apoptotic cells was performed via taking the images in random fields and counting at least 200 cells in four random fields in each well.
7. Quantification of MDC labeling of cells
The autofluorescent agent monodansylcadaverine (MDC; Sigma) was recently introduced as a specific autophagolysosome marker to analyze the autophagic process. HeLa cells were treated with paclitaxel 1 μg/mL for 24 hours. Autophagic vacuoles were labeled with MDC by incubating the cells with 0.05 mM MDC in DMEM at 37°C for 10 minutes, at various times after the paclitaxel treatment. After incubation, the cells were washed three times with PBS and immediately analyzed with a fluorescence microscope (Nikon Eclipse TE 300, Nikon, Tokyo, Japan) equipped with a filter system (V-2A excitation filter, 380/420 nm; barrier filter, 450 nm). Images were captured with a CCD camera and imported into Adobe Photoshop.
8. Statistical analysis
Statistical analysis of the results was performed by analysis of variance (ANOVA) and Tukey multiple range tests, using a Prism ver. 2.0 software (GraphPad Software, San Diego, CA). Statistical significance was defined as p ≤ 0.05.
Results
1. Autophagy was induced in HeLa cells treated with paclitaxel
An increasing number of studies have shown that autophagy is activated during stress conditions for the degradation of superfluous proteins and damaged organelles. In line with these studies, we observed that (as shown in Fig. 1), compared with the control cells, HeLa cells treated with paclitaxel showed an increase in the number of MDC labeled vesicles, suggesting an induction of autophagic vacuoles formation after paclitaxel treatment. Furthermore, autophagy related genes were up-regulated. Autophagy marker gene Beclin 1 and LC3 II were induced; however, p62 was downregulated by paclitaxel treatment (Fig. 2). The results indicated that autophagy was induced by paclitaxel exposure in HeLa cells.
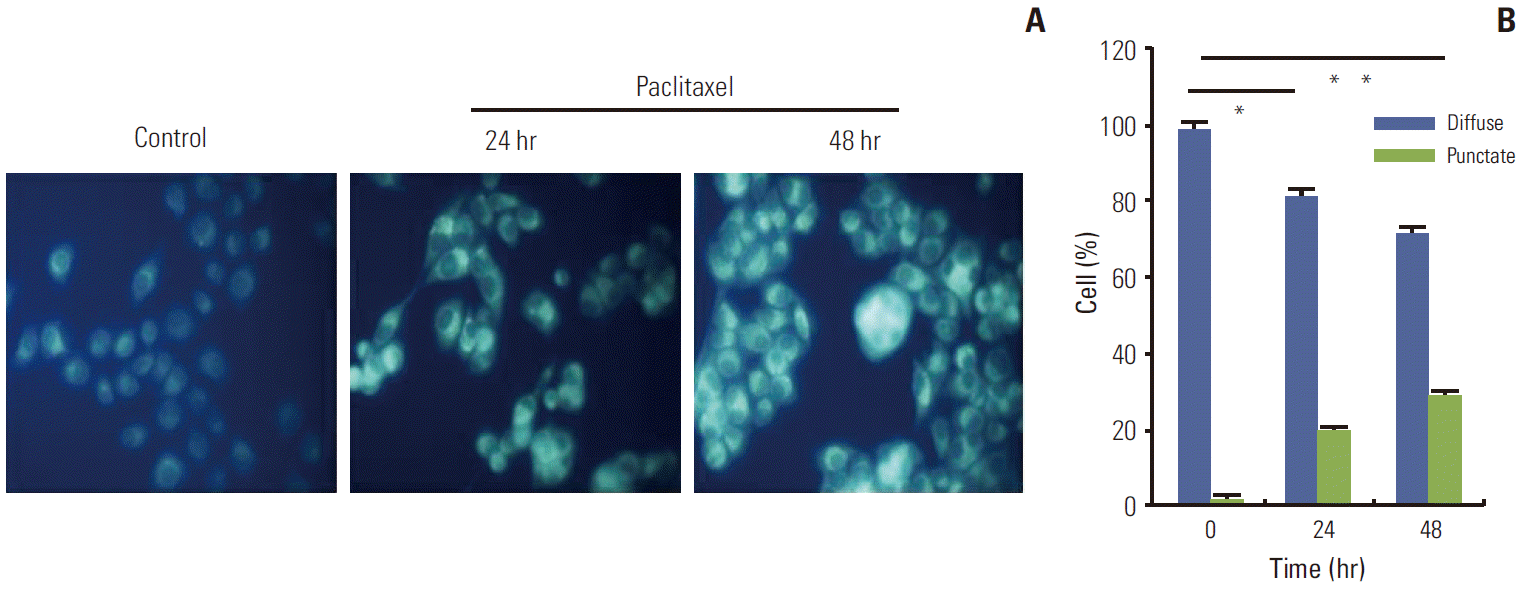
Monodansylcadaverine (MDC) labeled vesicles in HeLa cells treated with paclitaxel. HeLa cells were treated with paclitaxel 1 μg/mL for 24 hours. Autophagic vacuoles were labeled with MDC by incubating cells with 0.05 mM MDC in Dulbecco’s modified Eagle’s medium at 37°C for 10 minutes at various times after paclitaxel treatment. Compared with the control cells, HeLa cells treated with paclitaxel showed an increase in the number of MDC labeled vesicles, suggesting an induction of autophagic vacuoles formation after paclitaxel treatment. (A) The image of MDC labeled vesicles in HeLa cells. (B) The quantitative result of MDC labeled vesicles. *p < 0.05, **p < 0.01.

Detection of autophagy related genes expression by Western blotting in HeLa cells after treatment with paclitaxel for 24 hours and 48 hours. HeLa cells were treated with paclitaxel 1 μg/mL for 24 hours and 48 hours and then cell lysates were collected. Autophagy marker gene Beclin 1 and LC3 II were induced, but p62 was downregulated by paclitaxel treatment, indicating that autophagy was induced by paclitaxel exposure in HeLa cells.
2. Paclitaxel induced ER stress in HeLa cells
Recent literature suggests that disturbances in the integrity of ER activate autophagy [6]. To investigate the molecular mechanisms behind paclitaxel induced autophagy in HeLa cells, we investigated whether paclitaxel can trigger ER stress. To address this issue, HeLa cells treated with paclitaxel were detected by Western blotting. GRP78 is an ER chaperone protein, which is upregulated by ER stress. When the cells were treated with paclitaxel, GRP78 protein expression was significantly increased at 8 hours and 16 hours (Fig. 3). Our data further showed that paclitaxel induced ER stress was accompanied by the upregulation of PERK and IRE1 in HeLa cells. Both PERK and IRE1 are regulated by GRP78 during ER stress (Fig. 3). These observations indicated that paclitaxel can induce ER stress in HeLa cells.
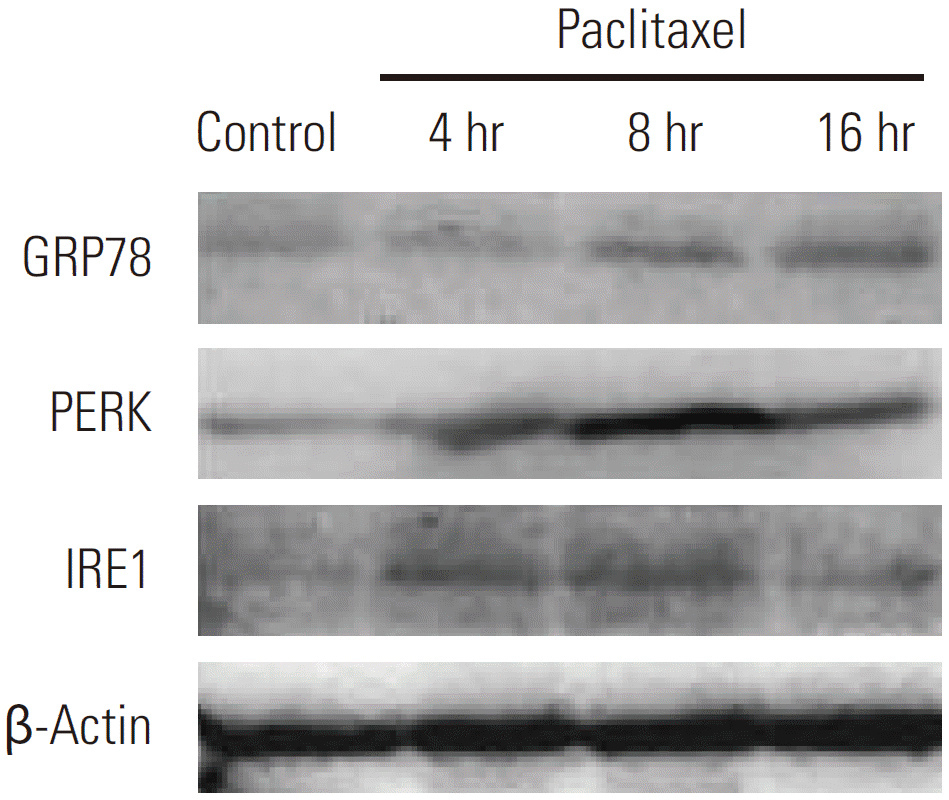
Detection of endoplasmic reticulum stress related genes expression by Western blotting in HeLa cells after treatment with paclitaxel. HeLa cells were treated with paclitaxel 1 μg/mL for 4 hours, 8 hours, and 16 hours and then cell lysates were collected. Glucose-regulated protein-78 (GRP78) protein expression was significantly increased at 8 hours and 16 hours, and the upregulation of PERK and IRE1 in HeLa cells were observed.
3. Blocking autophagy exacerbated UPR
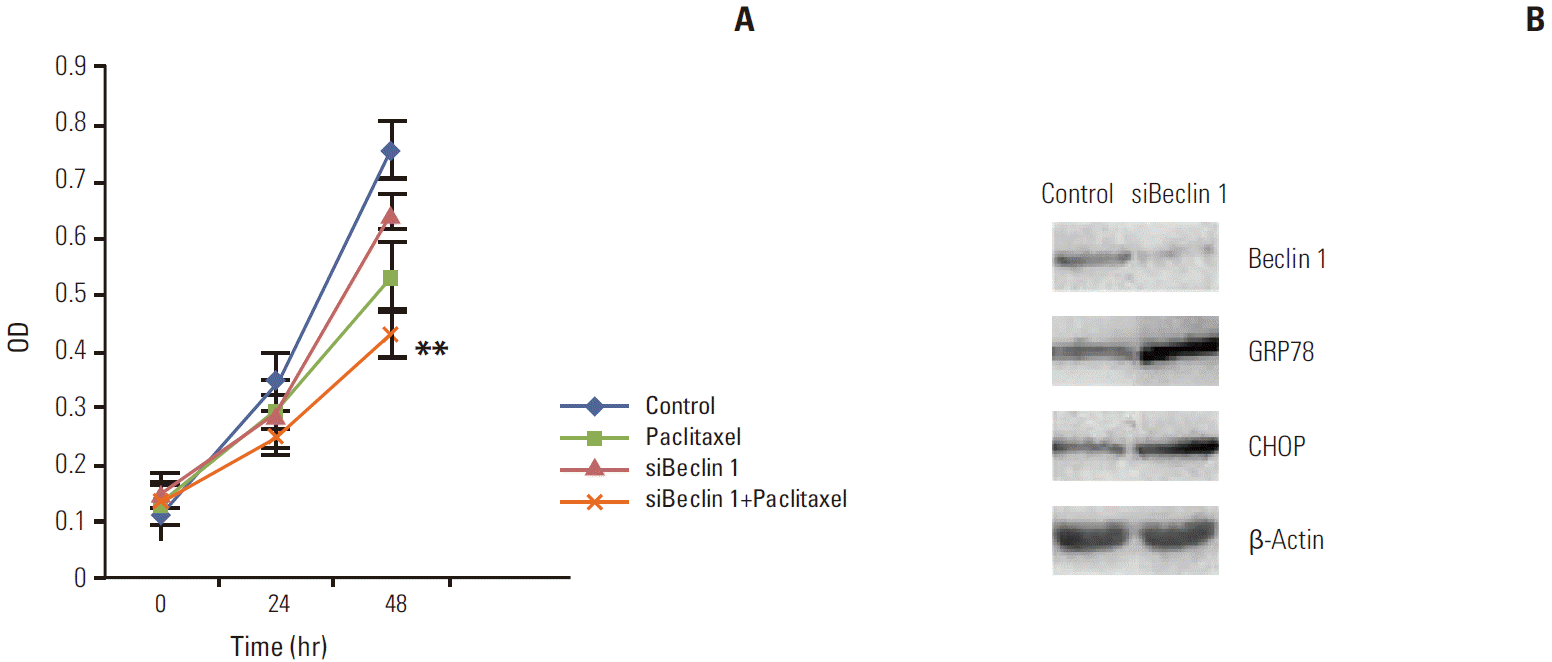
Since we observed that HeLa cells exposed to paclitaxel undergo both ER stress and autophagy, we hypothesized that autophagy in cervical cancer cells is activated by paclitaxel through an induction of ER stress. Firstly, we used autophagy pharmaceutical inhibitor CQ to treat HeLa cells after paclitaxel exposure to detect UPR related genes expression. Our results show that CQ administration, following paclitaxel treatment, predominantly induced the expression of p62 and blocked the increase in the expression of LC3B and Beclin 1, while elevating the expression of GRP78 and CHOP (as shown in Fig. 4). To firmly establish the effects of autophagy inhibition on ER stress in HeLa cells after paclitaxel treatment, Beclin 1 was transiently silenced using siRNA. Our results showed that the cytotoxic effect of paclitaxel was significantly increased by blocking Beclin 1 expression when compared to the random control (Appendix 1A). To gain further insight into this mechanism, we detected the expression of GRP78 and CHOP (Appendix 1A and B). As expected, the expression of GRP78 and CHOP were enhanced. These results support our hypothesis that paclitaxel could induce accumulation of unfolded or misfolded proteins, while autophagy could clear them to attenuate ER stress in HeLa cells.
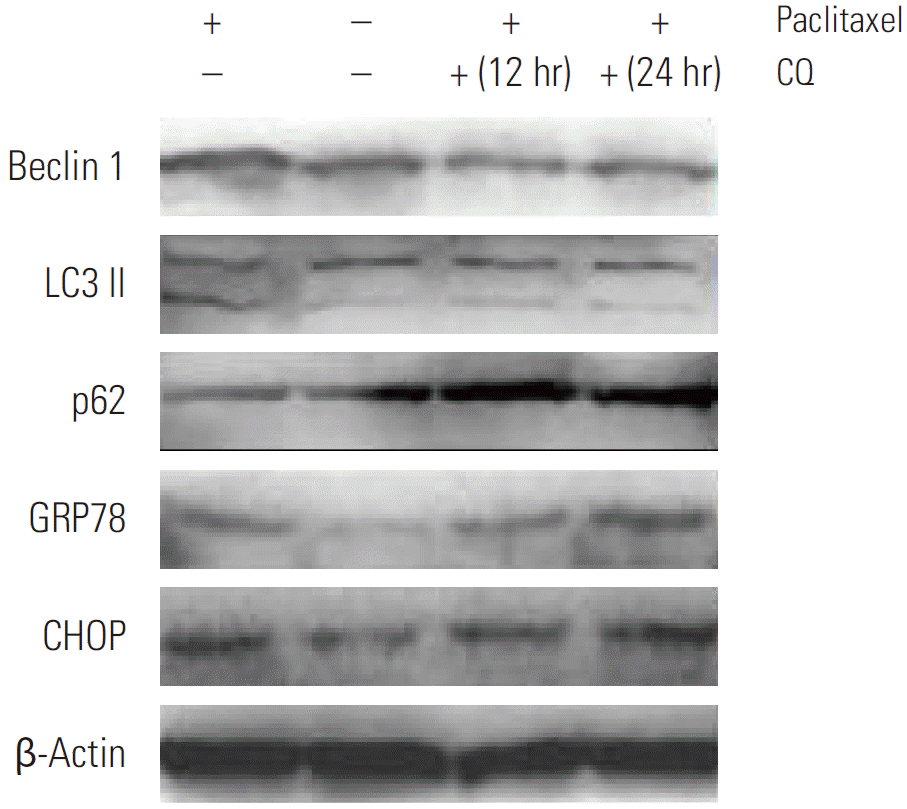
Detection of autophagy and endoplasmic reticulum stress related genes expression by Western blotting in HeLa cells after treatment with autophagy inhibitor chloroquine (CQ) for 12 hours and 24 hours following paclitaxel exposure. Firstly, HeLa cells were treated with paclitaxel 1 μg/mL for 2 hours, then treated with 5 μM CQ for 12 hours and 24 hours. CQ administration following paclitaxel treatment predominantly induced expression of p62, blocked the increase in the expression of LC3 II and Beclin 1, and meanwhile elevated the expression of glucose-regulated protein-78 (GRP78) and CHOP.
4. Inhibition of autophagy potentiates ER stress-mediated apoptotic cell death
Accumulation of unfolded or misfolded proteins in the ER lumen induced activation of chaperones and the recovery process. When the recovery process autophagy was inhibited, severe ER stress causes cell death, usually by an activation of intrinsic apoptosis. Therefore, we next observed the cell apoptosis ratio and tested the expression of ER stress related apoptosis genes after autophagy inhibition. As shown in Fig. 5, cell apoptosis induction by paclitaxel was markedly enhanced in the presence of CQ. Consistent with the results using CQ, the degree of apoptosis induced by paclitaxel was also enhanced when Beclin 1 protein level was reduced by a specific siRNA, as observed with Hoechest 33258 staining (Fig. 6). More apoptotic cells were observed in Beclin 1 siRNA–transfected cells after paclitaxel treatment than in the control cells treated with paclitaxel. These results demonstrate that autophagy induced by paclitaxel conferred protection of tumor cells against apoptosis, and blockade of autophagy subsequently enhanced the apoptosis associated with paclitaxel treatment.

Apoptosis ratio of HeLa cells treated with paclitaxel in the presence of chloroquine (CQ). HeLa cells were harvested and resuspended in binding buffer, after treatment with paclitaxel (1 μg/mL) for 24 hours or paclitaxel treatment followed by CQ (5 μM) treatment for 48 hours. Cell apoptosis were then analyzed by flow cytometry. The percentage of cells with a sub-G1 DNA content was taken as a measure of cell death. At least three independent experiments were performed. *p < 0.05, **p < 0.01. Cell apoptosis induction by paclitaxel was markedly enhanced in the presence of CQ. PI, propidium iodide; PCX, paclitaxel.

Hoechest 33258 staining of HeLa cells after different treatment. The cells were transfected with siRNA and/or treated with paclitaxel (1 μg/mL) for 24 hours. After a change of fresh medium, the cells were incubated with Hoechst 33258 solution followed by observation under a fluorescence microscope. Strong fluorescence can be observed in the nuclei of apoptotic cells, while weak fluorescence was observed in non-apoptotic cells. Quantification of apoptotic cells was performed by taking the images in random fields and counting at least 200 cells in four random fields in each well. More apoptotic cells were observed in Beclin 1 siRNA–transfected cells after paclitaxel treatment than in control cells treated with paclitaxel. PCX, paclitaxel. *p < 0.05, **p < 0.01.
Discussion
Cervical cancer is the most common malignancy of the female reproductive system. Although neoadjuvant chemotherapy, along with concurrent chemo- and radiotherapies have benefited majority patients, survival in women with recurrent or metastatic cervical cancer remains poor. Resistance of cancer to chemotherapy is one of the primary causes of treatment failure [6,7]. Paclitaxel is the prototypic member of the taxane class of mitotic inhibitor-based chemotherapies. Paclitaxel and its analogs are widely used in the treatment of solid tumors, including cervical cancer, especially in a combination with platinum [8]. Therefore, other therapeutic approaches must be tested in order to further improve the prognosis.
Many studied have shown that antitumor activity of paclitaxel is due to its interaction with b-tubulin, where it inhibits microtubule depolymerization and arrests cycling cells in the mitotic (M) phase. Finally, paclitaxel activates the apoptotic signal pathway, leading to caspase activation, which involves the Bcl-2 family of proteins and the mitochondria [9,10]. Recently, the ER was reported to be a cytosolic target of paclitaxel induced apoptosis via the ER stress pathway; sustained ER stress induces caspase-mediated apoptosis [11]. However, the exact mechanisms of paclitaxelinduced cell death are not completely illustrated.
ER is a central cell organelle that is involved in biosynthesis, protein folding, and modification of proteins [12]. ER is very sensitive to minor perturbations in its environment. A number of cellular stress conditions, such as glucose deprivation, oxidative stress, and infection, can lead to the accumulation of unfolded or misfolded proteins in the ER lumen, which disturbs the ER function and result in ER stress [13]. ER stress leads to the UPR to avoid cell damage, which triggers two downstream signaling events. One is the induction of genes encoding for ER resident chaperones to properly fold the unfolded proteins and the other being suppression of initiation of protein synthesis [14]. However, severe ER stress can cause cell death, usually by an activation of apoptosis [15]. Moreover, in order to clear the ER of accumulated terminally misfolded protein aggregates that cannot be degraded by the proteasome, the UPR may upregulate the autophagy machinery [16,17].
Autophagy plays an important physiological role in the turnover of cellular proteins and other macromolecules. Autophagy can be induced by a change in the environmental conditions, such as nutrient depletion; moreover, autophagy is involved in development, differentiation, and tissue remodeling in various organisms [18,19]. In addition, autophagy can also be induced in a response to chemotherapy and has been considered as an important mechanism contributing to drug resistance, and the autophagic process may protect cancer cells from toxicity [20]. Therefore, the inhibition of autophagy may result in a greater susceptibility to cell death-inducing mechanisms.
In our study, we hypothesized that inhibition of autophagy leads to aggravated ER stress, which then induces downstream apoptosis. We firstly detected the expression of autophagy marker genes Beclin 1, LC3II, and p62 in HeLa cells treatment with paclitaxel, and then detected the expression of ER stress marker genes GRP78 and CHOP. Although the results showed that the expression of these genes had been changed after paclitaxel treatment, a conclusion cannot be made since autophagy and ER stress are closely related. Then, we detected the expression of ER stress related genes in HeLa cells after the treatment with an inhibitor of autophagy or siRNA against the autophagy gene to confirm the relationship between autophagy and ER stress. The results demonstrated that a blockade of autophagy can increase the expression of the ER stress genes. Moreover, ER stress-mediated apoptotic cell death after paclitaxel treatment was potentiated by an inhibition of autophagy. These results preliminarily indicated that inhibition of autophagy led to the aggravation of ER stress and potentiated downstream cell apoptosis. CQ is a lysosomotropic drug that raises intralysosomal pH level and impairs autophagic protein degradation [21]. By blocking the last step of the autophagy pathway, CQ can increase protein accumulation in the cells more than 3-methyladenine (3MA) which is known to be a representative autophagy inhibitor by mainly blocking the initiation of autophagy pathway. To test our hypothesis, we used CQ but not 3MA to treat HeLa cells to induce protein accumulation; then detected the expression of the ER stress genes and cell apoptosis. The result showed that p62 was dramatically elevated after CQ treatment, which means protein degradation was significantly blocked and more protein accumulated. We also wanted to know whether the inhibition of the initiation of autophagy could aggravate ER stress as such, we used siRNA against Beclin 1 to test this. The results showed that either CQ or siRNA against Beclin 1 could promote the expression of ER stress genes and potentiate downstream cell apoptosis. Therefore, a blockade of autophagy could aggravate ER stress and promote cell apoptosis in HeLa cells.
Xi et al. [22] reported that paclitaxel can promote apoptotic cell death, accompanied with an induction of autophagy in lung cancer A549 cells. Kim et al. [23] also revealed that paclitaxel can increase autophagic activity, suppressing the autophagy enhancement of paclitaxel-induced apoptosis in OS cells. Conversely, the over-expression of Beclin1 in cervical cancer CaSki cells may enhance apoptosis signaling induced by anti-cancer drugs (cisplatin, paclitaxel, 5-fluorouracil, and epirubicin), which suggests that autophagy can promote apoptosis after drug treatment [7]. In mitotic cells, paclitaxel can block the activation of class III phosphatidyl inositol 3 kinase, Vps34, a critical initiator of autophagosome formation. Chemically or genetically blocking autophagosome formation diminished paclitaxel-induced cell death, suggesting that autophagosome accumulation sensitized the cells to paclitaxel toxicity [8]. Interestingly, in v-Ha-ras-transformed NIH 3T3 cells treated with paclitaxel, both autophagy and apoptosis act as cooperative partners to induce cell death [1]. Paclitaxel treatment induced cell death through autophagy or apoptosis or both is different in different cells. Furthermore, autophagy sometimes leads to cell death, while other times protects cells from cell death. In our study, protective autophagy was induced after paclitaxel treatment in HeLa cells.
In U937 cells, ER would seem to contribute to paclitaxelinduced apoptosis via both the early release of Ca2+ and the late amplification of mitochondria-mediated apoptotic signals [24]. Thapsigargin, a classic ER stress inducer, can increase the cell killing potency of paclitaxel or docetaxel by 10- to 12-fold in Human prostate cancer PC-3 cells. In addition, both the intrinsic and extrinsic apoptotic pathways were involved in the sensitization mechanisms. ER stress-apoptotic response is conducive to chemotherapeutic sensitization [25]. Our research reveals that the inhibition of autophagy can intensify ER stress apoptotic response and sensitivity to paclitaxel.
Conclusion
Our results preliminarily showed that the inhibition of autophagy can intensify ER stress and induce downstream cell apoptosis in HeLa cells. The main finding shown in our study is consistent with previous studies, but our results contribute new knowledge to understand the role of ER-stress related autophagy in cancer cells after taxane treatment.
Notes
Conflict of interest relevant to this article was not reported.
Acknowledgements
The research is supported by Key Science and Technology Program of Shanxi Province (Grant No. 20120313019-2), Foundation of Public Health Department of Shanxi (Grant No. 2011024).
References
Appendices
The effect of autophagy inhibition on endoplasmic reticulum stress. Firstly, HeLa cells were treated with paclitaxel 1 μg/mL for 2 hours, then transiently transfected with siRNA for another 24-hour and 48-hour culture. (A) Result of cell proliferation assay. The cytotoxic effect of paclitaxel was significantly increased by blocking Beclin 1 expression when compared to the control. (B) Result of Western blotting. The expression of glucose-regulated protein-78 (GRP78) and CHOP were enhanced when Beclin 1 expression was knock down.