Present Status and Problems on Molecular Targeted Therapy of Cancer
Article information
Abstract
Numerous clinical trials of molecular targeted drugs for cancer have been conducted, with remarkable results for certain drugs and accumulation of "negative data" causing a hitch in the development plan for some other compounds. Five recent issues and problems of molecular targeted therapies were discussed critically. Drug discovery and effects against driver mutations (activating mutations) and problems: possibility for circumventing inherent and acquired resistance with the aim of achieving radical cure. Synthetic lethality: reasonable patient selection in individualized treatment strategy. Response rate and progression-free survival improvement with or without overall survival benefit and enhancement of toxicity in bevacizumab therapy: best endpoints for the evaluation of effect of antiangiogenic therapy. Negative data on small-molecule targeted therapy, primarily vascular endothelial growth factor tyrosine kinase inhibitors: loose GO or NO-GO decision criteria for further development of new compounds in early clinical trials. Effect of immunotherapy: difficulty to verify by proof of principle study. We are faced to many questions for the development of efficient personalized therapy. Accumulation of scientific global preclinical and clinical evidences is essential to use these new therapeutic modalities for the improvement of oncologic health care.
Drug Discovery, against Driver Mutations(Activating Mutation) Effects and Problems
Drugs against small-molecule substances and antibodies which specifically control genes serving as a driving force for cancer growth, called activated mutant gene, have been demonstrated to afford the most distinct beneficial effect among the molecular target drugs currently under investigation [1]. This is because growth of cancer is dependent solely on the mutant genes.
These molecular targeted drugs include imatinib directed against breakpoint cluster region-abelson (BCR-ABL) for chronic myeloid leukemia (CML), imatinib directed against mutant c-kit for gastrointestinal stromal tumor (GIST), erlotinib and gefinitib directed against mutant epidermal growth factor receptor (EGFR) for non-small cell lung cancer (NSCLC), crizotinib directed against echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) for NSCLC, and vemurafenib directed against mutant B-type Raf kinase (BRAF) for melanoma. All of these drugs have been demonstrated to yield high response rates and to significantly prolong the disease-free survival, progression-free-survival (PFS) and overall survival (OS) when administered alone, and are hence generally accepted as standard treatments for the relevant disorders (Table 1) [2,3].

One of the issues needings to be explored is that while a significant prolongation of OS was demonstrated for imatinib and vemurafenib in randomized comparative studies with cytotoxic anticancer agents, no such prolongation of OS has been demonstrated for gefinitib or erlotinib in spite of their dramatic antitumor activity. Such discrepant results may be attributable to the molecular target drugs being administered to virtually all patients in the chemotherapy-treated group in a crossover design after the disease has progressed. In fact, a comparative study of vemurafenib versus dacarbazine (DTIC) conducted in patients with advanced melanoma using a protocol that prohibited crossover medications demonstrated a significantly prolonged OS in the vemurafenib-treated group (Table 2) [4]. This study design was called into question by The New York Times and other media in the United States from the ethical viewpoint (Fig. 1) [5]. However, such a study design is perhaps acceptable under circumstances where it is uncertain whether a drug actually produces a theoretically anticipated effect or not. In cases for gefinitib and erlotinib, on the other hand, various clinical trials were conducted at the outset of the clinical development of these drugs under the assumption that the target might be wild-type EGFR, and it became evident that patient populations with such clinical characteristics as adenocarcinomas, non-smokers and females were likely to benefit to some extent from the targeted therapies. In April and May, 2004, it was reported that the true target of these drugs was a mutant EGFR [6,7]. Under such a situation, it became extremely difficult to prohibit a crossover design in comparative studies in patients bearing mutant EGFR due to ethical reason.


Second problem will be frequency of each driver mutation. BCR-ABL gene translocation is noted in practically all CML patients. c-kit mutation in GIST, in which cases imatinib is effective, EGFR mutation in NSCLC, and BRAF mutation in melanoma are detected in considerable proportion such as > 90%, about 10-35%, and 40-60%, respectively of the patients [8-10]. On the other hand, the EML4-ALK fusion gene in NSCLC is found in only about 4% of patients with adenocarcinoma of the lung [11]. The frequency of the BRAF mutation in NSCLC is ≤2% [12]. If different driver mutations were discovered in only small fractions of patients in the future, individualized drug discovery and selection of patients may possibly become more and more complicated. Another important problem to be tackled will be up to what extent of market size contraction the researchers'/firm's will to discover drugs can be maintained.
Third issue will be circumvention of drug resistance to each drug. These therapeutic agents produce dramatic effects in most patients at the beginning of the treatment, however, there are some patients who do not respond at all even in the presence of a driver mutation (Resistance 1). It has been pointed out that this could be attributable to the problem in low specificity for the detection of mutations. Even among responders, the majority of patients may fail to achieve complete response, but still show partial response (Resistance 2). Furthermore, occurrence of secondary mutations, such as threonine 790 methionine (T790M) in mutant EGFR, and emergence of resistance through other mechanisms such as activation of another signal transduction pathways are also encountered (Resistance 3). Secondary mutations have also been reported in lung cancer with EML4-ALK fusion gene [13,14]. It is yet to be clarified in regard to Resistance 3, whether the secondary mutation may actually be induced by a drug or represents the result of selection of a sensitive cell population that responds to the drug (Table 3). In any event, radical cure cannot be expected even with sharply acting molecular targeted drugs, unless Resistances 1, 2 and 3 can be circumvented. It is considered an important future subject of research for clinicians to design appropriate combinations of molecular targeted drugs and cytotoxic chemotherapy or combinations of different molecular-targeted drugs. Comparative studies of EGFR-tyrosine kinase inhibitor (TKI)±cytotoxic chemotherapy regimens in non-selected cases of NSCLC have failed to confirm an add-on effect of EGFR-TKI, however, there is still the hope that improved efficacy might be obtained with such therapy in EGFR mutation-positive cases by this strategy. Because median survival time of EGFR-mutation positive patients improved to 27-30 months after the beginning of treatment by the sequential combination of EGFR-TKI+cytotoxic drugs. A recent report described that irreversible EGFR-TKI+cetuximab was effective in EGFR mutation-positive patients who were no longer responsive to erlotinib or gefinitib. There is great hope, of course, that drugs capable of circumventing emergence of resistance will be discovered via elucidation of the underlying mechanisms [15,16].

Problems for cure by molecular target therapy
Patient Selection in a Synthetic Lethality Strategy
Synthetic lethality is a newly-coined term and has been used, for example, to explain how poly ADR-ribose polymerase (PARP) inhibitors are effective only in breast cancer susceptibility gene (BRCA) 1/2 mutation-positive cases [17]. PARP plays an important role in repairing single-stranded DNA cleavage at the sites of DNA damage caused by anticancer agents or radiation. Following administration of PARP inhibitors, the damaged DNA cannot be repaired, leading to accumulation of single-stranded DNA breaks which eventually result in double-stranded DNA breaks. Damaged DNA can be repaired by homologous recombination in cells with normal BRCA1/2, whereas in BRCA1/2-defective cells, no such repair of DNA damage is possible, resulting in cellular death [18-20]. Patients with ovarian cancer or breast cancer with BRCA 1/2 mutation/defect have been reported, and clinical studies of profound interest aimed at synthetic lethality are ongoing (Fig. 2) [18,19,21].

Mechanism for synthetic lethality in BRCA1/2 deficient cancer. Poly ADR-ribose polymerase (PARP) inhibition produces tumor-selective synthetic lethaity. When PARP action is inhibited, SS are converted to DSB at replication. In cells with functional HR pathway, the DSB will be repaired. In cells with a dysfunctional HR pathway, as is the case with BRCA 1 and 2, the lesions go unrepaired and cell death ensues [18,19,21]. BRCA, breast cancer susceptibility gene; DSB, double-strand break; HR, homologous recombination; SSB, single-strand break.
A comparative phase II clinical trial was conducted to assess maintenance therapy with olaparib (PARP inhibitor) in BRCA 1/2 mutation-positive patients after induction chemotherapy for advanced ovarian carcinoma. The study population comprised patients who had received two or more combination chemotherapy regimens including platinum. Although the study was a phase II trial, the results showed significant prolongation of the PFS (p < 0.00001) in patients treated with olaparib as a maintenance therapy [22]. The results of the phase II study performed in 265 patients with sensitive relapse encouraged to do next phase III study.
Triple-negative breast cancer (TNBC) bears similarity to hereditary BRCA1/2 mutation-positive breast cancer with respect to characteristics such as the estrogen-receptor/progesterone receptor/human epidermal growth factor receptor type 2 (ER/PR/HER2) status, p53 status, mutant-type gene expression pattern (basal like), histologic type (poorly differentiated), and sensitivity to DNA-damaging anticancer agents (highly sensitive). Diminished expression of BRCA1/2 is also noted. TNBC has been recognized to have a distribution which overlaps in part with that of BRCA1/2 mutation (defect)-positive breast cancer.
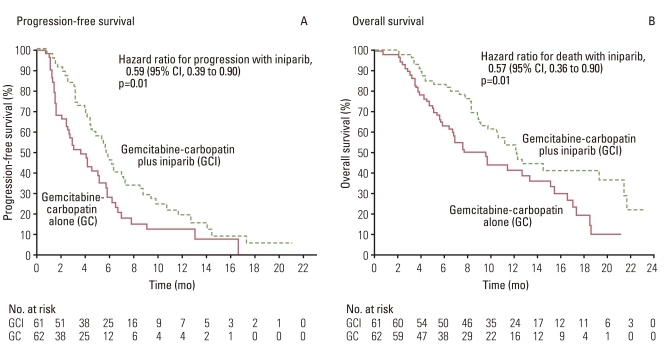
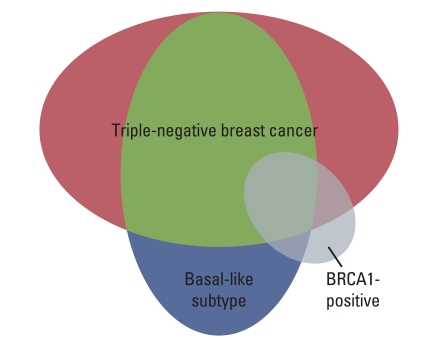
O'Shaughnessy et al. [23] conducted a comparative phase II study of gemcitabine+carboplatin±iniparib in 120 patients with TNBC and published the results in the New England Journal of Medicine. The iniparib add-on group showed both superior PFS and OS in this phase II trial (Fig. 3). With the aim of confirming the outcome, a phase III trial was undertaken in 519 previously treated patients with TNBC, using PFS and OS as the primary endpoints. The trial seemed a highly hopeful comparative study in which 519 TNBC patients were enrolled within a period of as short as 9 months from July 2009 to March 2010. The target number of recruited patients was set at 420 before starting the trial, with hazard ratios of 0.66 and 0.65 for OS and PFS, respectively, and α-error levels of 0.04 and 0.01, respectively, based on a previous phase II trial which had demonstrated an add-on effect of the PARP inhibitor. In the phase III study, however, the hazard ratios for OS and PFS were 0.88 and 0.79, respectively, with p-values of 0.28 and 0.027; hence neither endpoint cleared the estimated value, yielding negative data [24]. The results were one of the most disappointing among those of clinical studies on molecular targeted drugs reported in 2011 (Table 4) [24]. Most probable explanation for the disappointing data may be confusion of the concept of BRCA1/2 mutation (defect) with that of BRCA-ness. The background for the study population consisting of TNBC patients in clinical trials using a PARP inhibitor is based on the hypothesis of BRCA-ness that the BRCA1 promoter is methylated or the BRCA1 function is depressed in TNBC. In other words, mutation/defect of BRCA1/2 and BRCA-ness are entirely different concepts. As can be seen in the Fig. 4, TNBC, basal-like breast cancer (BC) and BRCA1-mutant or defective BC overlap for a considerable part, but BRCA1-mutant or defective BC may constitutes a small proportion of TNBC and basal-like BC [25]. Although, in the phase III trial, clinical characteristics in PR/ER/HER2-negative cases of TNBC were assessed, whereas there were no data concerning the BRCA1/2 status, it would be possible to infer that the proportion of BRCA1/2-mutant (defective) BC cases in the population enrolled in the phase II and III studies affected the study results. Thus, effects of the treatment cannot be expected in studies involving contradiction between the patient inclusion criteria and the target for the molecular targeted drugs. This suggests the cardinal importance of appropriate patient selection based on reliable biomarkers.

Response Rate and PFS Improvement with/without OS Prolongation and Enhancement of Toxicity of Bevacizumab (BEV) Therapy
Angiogenesis inhibitors have been evaluated in clinical studies on a variety of malignancies because they are considered to be non-organ-specific from the viewpoint of efficacy, in as much as angiogenesis inhibitors exert their effects on the cancer milieu rather than on the cancer cells per se. Biomarkers to estimate therapeutic responses, if present at all, cannot be used for a qualitative estimation of the responses to angiogenesis inhibitors in terms of a 'yes' or 'no.' Several comparative studies of BEV have been conducted, which have generated data indicating that while the response rate and PFS improved, there was still no significant improvement of the OS following BEV therapy combined with chemotherapy in most instances (Tables 5 and 6) [26]. Prolongation of OS occurred only in colorectal cancer, following use of BEV in combination with irinote can/fluorouracil/leucovorin or oxaliplatin/fluorouracil/leucovorin (FOLFOX) therapy and paclitaxel+carboplatin regimens for non-small cell carcinoma. In all studies including E2100 [28,42], BEV and Docetaxel (AVADO) [29] and Regimens in Bevacizumab for Breast Oncology (RIBBON) [30,43] trials conducted in patients with advanced BC, only the response rate and PFS improved, with no significant prolongation of the OS, therefore, cancellation of the accelerated approval on the basis of the E2100 trial data has been decided in United States Food and Drug Administration (US FDA) based on Oncology Drug Advisory Committee (ODAC) recommendation (Table 7). Similar results were obtained in the Gynecologic Oncology Group (GOG) 0218 [39], a randomized, double blinded, placebo-controlled, Phase III trial of chemotherapy with or without bevacizumab in patients with platinum sensitive recurrent epithelial ovarian, primary peritoneal or fallopian, tube cancer (OCEANS) [41] and a randomized, two arm, multicenter Gynecologic Cancer InterGroup trial of adding bevacizumab to standard chemotherapy (ICON7) [40] studies conducted in patients with ovarian cancer. A comparative phase II trial was performed in lung cancer in Japan using the same schedule as that in the E4599 study and this trial failed to demonstrate any significant improvement of the OS in the BEV treatment group. Possible reasons for these failures may include 1) the problem of bias in evaluating the PFS, namely, the PFS in the BEV-treated group merely appeared to be better at a glance due to observation bias; 2) an insufficient degree of improvement for antitumor effect of the chemotherapy prescribed concomitantly with BEV; and 3) alterations of the biological characteristics of the residual tumor after combined treatment with BEV-chemotherapy, which may cause the malignant cancer cells to survive and proliferate rapidly in spite of continued treatment. Issues that need to be investigated in relation to the use of BEV may include 1) validity of continued use of BEV even after completion of chemotherapy in combined treatment with chemotherapy; 2) non-negligible adverse reactions that are observed even after exclusion of high-risk patients; 3) high drug prices (Fig. 5); and 4) limited degree of efficacy. BEV is prescribed in only about 10 to 20% of patients, even in the United States, where it is approved for use in the treatment of non-small-cell carcinoma of the lung, therefore, the propriety of the use of BEV should be judged after sufficient analysis of various factors.

Clinical benefit with bevacizumab (BV) in phase III studies: part 1

Clinical benefit with bevacizumab (BV) in phase III studies: part 2

Voting results of ODAC for bevacizumab in breast cancer
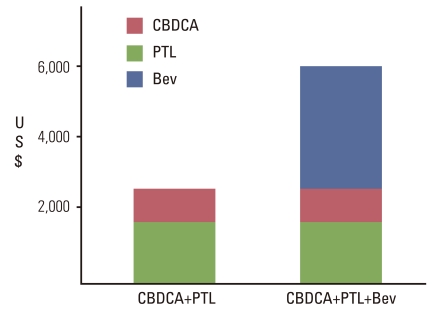
Increased treatment cost by bevacizumab (Bev). CBDCA, carboplatin; PTL, paclitaxel.
Accumulating Negative Data on Small-Molecule Kinase Inhibitors: Loose GO or NO-GO Decision Criteria on Further Development of New Compounds
Numerous studies for the development of small-molecule kinase (SMK) inhibitors are currently in progress. Among the most prominent SMK inhibitors that are under development are the multitargeted kinase inhibitors, targeting mainly on vascular endothelial growth factor receptor. Such multitargeted SMK inhibitors as sunitinib and sorafenib have been already in the market and are used as therapeutic agents renal cancer and hepatoma, but the precise mechanism of antitumor effect of these drugs remains unclear. The results of many clinical studies on SMK inhibitors under development have yielded negative data. The reason thereof may be said to be a mistake based on loose criteria for the GO or NO-GO decision about proceeding from non-clinical studies to clinical trials and from early-phase clinical trials to phase III trials [26]. It seems often the case that the magnitude of the antitumor effect is misjudged for data of non clinical studies. The non-clinical study data which have come to our notice are largely favorable and purport to demonstrate theoretically tenable antitumor effects. We must not disregard the possibility that there actually exists a vast amount of negative data underneath of positive results. On the other hand, insufficiently explained positive data also exist, and in some cases, an effect on a target distinct from an initially anticipated target is ultimately linked with the observed therapeutic effect. Designs of early phase clinical trials and interpretation of proof of principle (POP) data also have great influence on the GO/NO-GO decision preceding a big scale phase III trial. It is also an important subject as to which of the hitherto obtained study data recognized as global standards can be considered as reliable. Gefitinib and erlotinib are thought to be first-line drugs for the treatment of non-small-cell carcinoma expressing EGFR mutations; however, strangely, patient selection is generally considered unnecessary while prescribing erlotinib, as second-line treatment. Erlotinib has been strangely believed to be active even for patients without EGFR mutation in western investigators based on the results of BR-21 study [44]. On the other hand, gefitinib is considered to be inactive based on the results of IRESSAPan Asia Study (IPASS) trial [45] in n spite of the statistical demonstration for non-inferiority of gefitinib in Gefitinib and docetaxel trial in previously treated non-small cell lung cancer (INTEREST) study [46]. It would also be a future important subject to explore the validity of assessing the add-on effect of a new SMK inhibitor in a phase III comparative study of second-line and third-line regimens using erlotinib [47] in a comparator group in non-selected patients with non-small-cell carcinoma. It can be readily assumed that a comparative study of a multitargeted SMK inhibitor with or without a concomitant merely cytotoxic anticancer agent with the anticancer agent alone will yield negative data if tumor shrinkage may not be expected from monotherapy with the multitargeted SMK inhibitor directed towards mutations other than the driver mutation. This is simply because add-on effect can hardly be expected from a compound with only a small degree of antitumor effect.
Effect of Immunotherapy nearly Impossible to Verify by a POP Study
Immunotherapy was once regarded as a dubious treatment. Although the underlying mechanism of action can be cogently described, it is too complex and nearly impossible to verify through POP studies. There is no other means than demonstrating an overwhelming therapeutic effect of immunotherapy in scientific clinical studies to convince researchers, regulatory authorities and patients, in particular, under such circumstances. Recently, positive data on immunotherapy have been reported sporadically seen in the literature. Manufacture of Sipuleucel-T has been approved by the US FDA. Sipuleucel-T is a treatment consisting of injection of autologous peripheral blood dendritic cells, cultured in the presence of PA2024, a fusion protein composed of prostatic acid phosphatase (PAP) linked to granulocyte-macrophage colony-stimulating factor, which is an activated immune cell preparation targeted against PAP antigen. This complicated therapy was evaluated in a randomized placebo-controlled double-blind trial in 1,512 patients with asymptomatic, metastasis-positive, non-androgen-dependent cancer of the prostate (IMPACT Study). The mean survival time was 25.8 months in the Sipuleucel-T therapy group, as compared to 21.7 months in the placebo group (p=0.017); hence, the outcome was superior in the former [48], and the therapy was approved by the FDA on April 29, 2010, based on these results. This is a first approval of immunotherapy in US FDA.
At the 2010 American Society of Clinical Oncology (ASCO) Annual Meeting, a three-group parallel trial of the following three treatment groups, namely, ipilimumab+gp100, ipilimumab+placebo and gp100+placebo, in previously treated patients with malignant melanoma (molecular diagnostics [MDX] 010-20 Study) was reported. Gp100 antigen is expressed in the majority of melanoma cells. It has been reported that the cytotoxic T-lymphocyte antigen (CTLA)-4 on T cells inhibits binding of cluster of differentiation 28 (CD28) to B7 on the T cells by binding B7 on antigen presentation cells, thereby suppressing T cell activation (Fig. 6). It has been speculated that the binding of CD28 to B7 becomes possible when CTLA-4 is blocked by ipilimumab, resulting in T cell activation, and its function becomes enhanced. In the MDX010-20 Study with the primary endpoint consisting of assessments of the prolonging effects for OS of the ipilimumab+gp100 group (Arm A) and gp100 (Arm C) as the positive control group, the study was designed to also enable comparison of the gp100 group and an ipilimumab group (Arm B) (Table 8). Results of the study showed that the results in Arms A and B were significantly superior in terms of the OS than in Arm C. There was no significant difference between Arms A and B in the OS, although it was a little bit better in Arm B [49]. At the 2011 ASCO Annual Meeting, a randomized comparative trial to evaluate the add-on effect of ipilimumab to dacarbazine (DTIC), which is the standard treatment for previously untreated melanoma was reported. The combined therapy of ipilimumab and DTIC group showed superior OS (hazard ratio, 0.72; p=0.0009), PFS and duration of sustained response (Table 9) [50]. It seems very likely that this combination therapy will become the standard treatment for melanomas, and studies may probably generate results of various assessments of this treatment as adjuvant therapy.
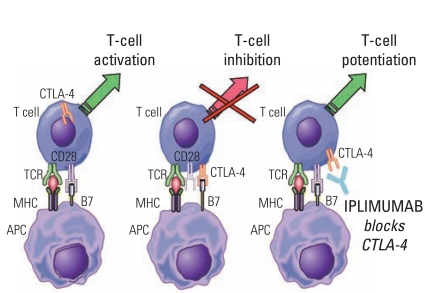
Mode of action of ipilimumab. CTLA-4, cytotoxic T-lymphocyte antigen-4; TCR, T-cell receptor; MHC, major histocompatibility complex; APC, antigen-presenting cell.


Ipilimumab is an antibody to CTLA-4 and works by restoring T cell activity. It has the effect of indirectly potentiating immunity, yet the effect is non-specific. It is considered an important problem to deal with how POP studies should be conducted in the case of such a compound. Rapid improvement of therapeutic responses using combined vemurafenib plus ipilimumab therapy for BRAF V600E mutation positive melanoma is expected.
Conclusion
Five topics pertaining to the effects and problems of the currently available molecular targeted therapies for malignancies have been reviewed critically. Personalized cancer therapy using molecular targeted drugs have just integrated in clinical practice, however, majority of cancer patients still cannot be cured. Beside, molecular targeted therapies show more than minimal problems including patient selection, drug induced adverse events and high costs. Physicians should make an effort to be fully aware of these problems and to devote for research and medical practice to improve oncologic medical care.
Notes
Conflict of interest relevant to this article was not reported.