INTRODUCTION
NSCLC is a worldwide common malignancy, and one of the leading causes of cancer related death. On initial diagnosis, 60 to 70% of patients with NSCLC have a locally advanced or metastatic disease (1). Patients with advanced disease who receive best supportive care survive for a few months, and approximately 90% survive less than 1 year (2).
Recently, the use of chemotherapy for advanced NSCLC has become more customary. Randomized trials have also shown that cisplatin-based chemotherapy and new agents result in an improved quality of life compared to the best supportive care (3). Further more, meta-analyses have demonstrated that, compared to supportive care, chemotherapy results in an improvement in survival, symptoms and quality of life for patients with advanced NSCLC (4~7). A number of new agents have been introduced over the last decade for the treatment of NSCLC, including paclitaxel, docetaxel, gemcitabine, vinorelbine, irinotecan, tirapazamine and carboplatin. Platinum-based combination therapy is currently considered the most active treatment option for patients with locally advanced or metastatic NSCLC (8~10). The Eastern Cooperative Oncology Group (ECOG) reported results of a randomized clinical trial to compare the efficacy of four commonly used regimens. This trial showed improved survivals at one and two years, but no significant difference in the survival was observed among the four regimens (10).
Gemcitabine, as a single-agent therapy, has been studied extensively and shown response rates of approximately 20% in patients with chemotherapy-naive advanced NSCLC (11~14). Gemcitabine has also shown preclinical and clinical evidence of synergism with cisplatin, with remission rates of 26 to 65.3% in phase II trials (15~21).
Based on these studies, a phase II clinical trial was conducted to evaluate the efficacy and toxicities of the combination regimen of gemcitabine and cisplatin as a first-line treatment in patients with advanced NSCLC.
MATERIALS AND METHODS
1) Eligibility
Patients with pathologically proven stage IIIB with malignant pleural or pericardial effusion or stage IV NSCLC, were enrolled in our trial. The patients were required to be at least 18 years of age, with an Eastern Cooperative Group (ECOG) performance status of 0 to 2 and a life expectancy of at least 12 weeks. The presence of at least a unidimensional measurable disease was mandatory, but a bidimensionally measurable disease was preferable. Additional eligibility criteria included: adequate bone marrow reserve (white blood cell count ≥3×109/L, platelets ≥100×109/L, hemoglobin ≥10 g/L, and hematocrit ≥30%) and liver and renal functions (creatinine < 1.5 times of the upper normal limit).
Patients were excluded from the study if they had an active infection, uncontrolled CNS metastasis requiring emergency radiotherapy and/or corticosteroids, serious concomitant systemic disorders, second primary malignancy (except in situ carcinoma of the cervix or nonmelanomatous skin cancers) and severe cardiovascular diseases. Patients who were pregnant or breast-feeding were not entered onto the study. All patients gave their informed consent prior to study enrollment.
2) Pretreatment evaluation
Before the initiation of chemotherapy, all patients underwent a complete medical history and physical examination, determination of performance status and measurement for a bidimensionally measurable disease. A complete blood cell count, biochemical tests and chest X-ray were performed before each cycle of therapy. Patients were followed up with measurements of the objective tumor parameters at the baseline, and after 2 and 4 cycles of treatment.
3) Treatment plan
Patients received cisplatin, 75 mg/m2, on day 1 as a 30 minutes intravenous infusion, with adequate hydration, diuretics and mannitol, followed by gemcitabine, 1,250 mg/m2, over 30 minutes on days 1 and 8 of each cycle. Also, a standard antiemetic regimen, with 5-HT3-antagonist, was administered. Treatment was repeated every 3 weeks. If the patient did not demonstrate progression of the disease, the treatment was continued for up to a total of six cycles. If the patient demonstrated a response, the treatment was continued at the physician's discretion.
4) Dose adjustments and evaluation during treatment
In the event of hematological and severe non-hematological toxicities occurring at any time during treatment, patients were treated by adjusting the dose of chemotherapeutic agents. With regard to the hematological toxicity, a complete blood cell count with differential and platelet counts were performed on each day of treatment. If grades 2 or 3 neutropenia/thrombocytopenia were observed on day 8, the dosage of gemcitabine was reduced to 75%. If grade 4 hematological toxicities occurred, the gemcitabine treatment was omitted. Patients that developed either neutropenic fever requiring antibiotic therapy or bleeding associated with thrombocytopenia received 75% of both the cisplatin and gemcitabine dosages in the subsequent cycle. A cycle was not started until the absolute neutrophils and platelet counts were greater than 1.5×109 and 100×109/L, respectively. For grade 3 non-hematological toxicities, patients received 75% of the gemcitabine dose. With grade 3 renal toxicity, treatment was withheld until recovery, and grade 2 the dose of cisplatin was reduced to 80%. Treatment was delayed after a maximum of 14 days. The toxicity profiles were graded according to the National Cancer Institute (NCI) common toxicity criteria version 2.0.
5) Determination of response
Responses were assessed at the end of every other treatment cycle according to the World Health Organization (WHO) criteria. A complete response (CR) was defined as the complete disappearance of all known disease, without the appearance of new lesions, confirmed by two examinations performed at least 4 weeks apart. A partial response (PR) was defined as a decrease of 50% or more in the sum of the products of the perpendicular diameters of each measurable lesion, confirmed by two examinations performed at least 4 weeks apart, with no progression or appearance of other and new lesions. A disease that failed to fulfill the above criteria, but did not show a 25% or greater increase in the total tumor load, was classified as a stable disease (SD) in the absence of any new lesions. A progression of disease (PD) was defined as an increase of 29% or more in the size of at least one measurable lesion, the appearance of new lesion (s) and/or the onset of ascites of pleural effusion with cytological confirmation.
6) Statistics
The duration of an objective (complete or partial) response was measured from the time the response was first documented to the date of disease progression. Overall survival (OS) was measured from the date of the initial treatment to the date of death. Time to progression (TTP) was defined as the period from the time of treatment to that of disease progression or discontinuation due to death or drug-related toxicities. The response rates, according to the prognostic factors, were compared by the Fisher's exact test. The survival curves were calculated using the Kaplan-Meier method. All parameters were calculated at a 95% CI. All analyses were performed using the SPSS software version 10.0 (SPSS, Inc, Chicago, IL). The level of significance was set at p≤0.05.
RESULTS
1) Patients characteristics
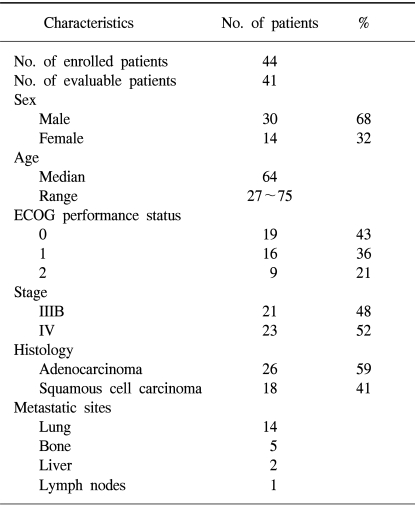
Between February 2001 and October 2002, 44 patients, 30 men and 14 women, with a median age of 64 years, ranged from 27 to 75 years were enrolled in the study. Of all the patients, 80% had an ECOG PS of 0 or 1, and 52% had a stage IV disease. Most patients had an adenocarcinoma (59%). These characteristics are listed in Table 1.
2) Response evaluation
Of the 44 patients, 41 were evaluable for response. Twenty two patients showed a PR, without CR, so the overall response rate (ORR) was 53.6%. Four patients (9.7%) showed a SD. By an Intent-to-treatment analysis, the objective response was shown to be 50%. No significant differences in response rates according to age, gender, stage, performance status and histological type were observed.
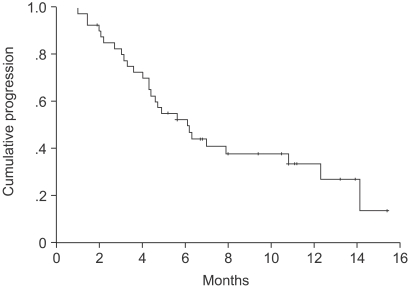
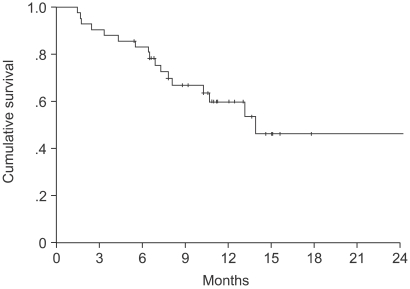
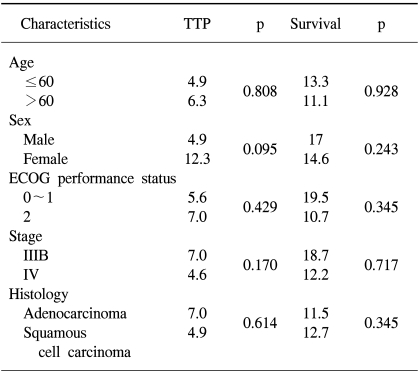
The median TTP was 6.1 months (95% CI, 4.1~8.1 months) (Fig. 1). Log-rank tests were applied for intergroup comparisons according to age, gender, stage, ECOG PS and histological type. However, no statistically significant differences were shown between the groups (Table 2). The overall median survival time and 1-year survival rate for all patients were 14.2 months (95% CI, 13.8~22.5 months) (Fig. 2) and 55% respectively. From a log-rank test for survival, no statistically significant differences were observed (Table 2). The median follow-up duration was 9.4 months.
3) Toxicity
A total of 179 cycles were evaluable for toxicity, and the incidences of toxicities are summarized in table 3. The most common hematological toxicity was thrombocytopenia, and there were no treatment-related mortalities.
DISCUSSION
In this phase II study, the combination regimen of gemcitabine and cisplatin in advanced NSCLC was shown to be highly effective and safe. The response rate was 53.6%, and 9.7% of patients were also observed to achieve stabilization. The median TTP was 6.1 months.
Many phase II trials have been conducted using different dosages and schedules on the combination of gemcitabine and cisplatin for patients with advanced NSCLC. Variable response rates, from 26 to 65.3%, have been with TTP from 4.2 to 9.9 months (15~22). A recently published Korean phase II trial of gemcitabine and cisplatin combination therapy showed an ORR and median survival of 47.8% and 11.8 months, respectively, for patients with advanced NSCLC (22). The recent phase III trial of EORTC 08975 indicated an ORR and median TTP of 36.8% and 7.4 months, respectively, for the combination of gemcitabine with cisplatin (23). In a comparative study of four regimens for advanced NSCLC by the ECOG, the combination of gemcitabine and cisplatin offer an ORR, median OS, 1- and 2-year survival rates and a median TTP of 22%, 8.1 months, 36 and 13% and 4.2 months, respectively. Although none of the four chemotherapy regimens offered significant response rate and survival advantages, the median TTP of the gemcitabine and cisplatin combination was statistically higher than the other regimens (10). Based on these similar efficacies, other parameters may become important, such as the toxicity, convenience and cost of the regimen. Although our study failed to demonstrate significant differences according to the prognostic factors, it did show relatively better ORR, TTP and survival than the other studies. The major reason for this result seems to be the higher proportion of patients with good performance status and a stage IIIB disease. If further studies are conducted with an adequate sample size, long term follow-up and evaluation of the quality of life, more positive results, with correlation to the prognostic factors, should be acquired.
This study demonstrated that the combination of gemcitabine and cisplatin had feasible and manageable toxicities. The most common hematological toxicities were thrombocytopenia (55.2%) and anemia (46.6%). The most common non-hematological toxicities were fatigue (42.4%) and neuropathy (25.2%), but these were limited to grade 1/2. These toxicity profiles were similar to those of the other studies. Unexpectedly, Pruritus was observed in 5.1% of our study group, which was usually limited to the extremities, with low-grade intensity. This toxicity was thought to be a dermatologic adverse event due to the gemcitabine. The overall tolerance of this treatment seems to be acceptable and compatible, as shown by other studies.
More recent phase III trials indicate that a therapeutic plateau may have been reached with conventional chemotherapy. Therefore, new molecular targeted agents have been incorporated into active chemotherapy regimens. However, promising results are yet to be obtained. Research on a molecular basis for lung cancer should be performed to improve the survival and overcome therapeutic plateau of conventional chemotherapy in patients with lung cancer.