INTRODUCTION
Radiation therapy is a general curative as well as palliative treatment mode for early and advanced uterine cervical cancer. A number of reports describe the effects of radiation on normal and cancerous tissues. Radiation mediates tumor growth in several ways, such as the induction of DNA damage, effects on cell membranes, activation of signal transduction pathways in cells, induction of the expression of immediate and late genes and impairment of cell cycle, etc (1~4). The sensitivity of mammalian tumor cells to radiation varies, even within the same type of cancer. It has been reported that differences in sensitivity are determined by the specific sensitivity of cells to radiation and the ability of these cells to recover after exposure to radiation (5). Hence, to enable effective treatment of tumors with reduced specific sensitivity to radiation, efforts have been made by numerous investigators to characterize radiation responding factors in cells (6). However, as the radiation-induced specific intracellular targets and relevant signal transduction pathways remain to be elucidated, further research into the genetic response of cells to radiation is required to improvement the efficacy of radiation therapy.
Investigations into the effects of radiation have progressed more rapidly since the development of molecular biological analytical methods (7,8). It is anticipated that the application of molecular biological methods to radiation therapy will tremendously improve its effectiveness, while simultaneously reducing its side effects.
Methods for assessing gene expression, such as Northern blot analyses, RNase protection assays, semiquantitative reverse transcription polymerase chain reaction (RT-PCR) and competitive RT-PCR have been widely used (9). However, the numerous disadvantages of these methods, such as low sensitivity, a long assay time, high rates of contamination and inaccurate outcomes, etc., mean that they have some experimental limitations. In 1992, to overcome some of the disadvantages of conventional PCR, Higuchi et al. developed a method that quantified amplified double strand DNA through staining with ethidium bromide, illumination with UV and assessment of the generated fluorescence with a CCD camera (10). However, the accuracy of this method is reduced in situations where the fluorescence is generated non-specifically. Real-time quantitative RT-PCR methods have overcome this problem, because they quantify the concentrations of amplified gene fragments by the incorporation of fluorescent substances into the gene-specific primers. This is currently the best available method for rapid, real-time quantification of gene expressions.
To characterize genes in HeLa cells, derived from uterine cervical cancer, which were differentially expressed in response to radiation, suppression subtraction hybridization (SSH) was performed. Several hundred clones were obtained, and approximately 30 candidate genes selected by reverse Northern blot analysis and their HIF-responsive gene, RTP801, identified. Using a real-time quantitative RT-PCR assay, the RTP801 was confirmed to be differentially expressed in response to radiation.
EXPERIMENTAL METHODS
1) Cells
Experiments were performed using stationary phase HeLa cells. Cells were cultured with DMEM containing 10% fetal bovine serum (FBS) and 1x antibiotics, Pen/Strep Fungizone Mixture (Cambrex Bio Science Walkersville, Inc. MD, USA). Experimental cells were irradiated, then removed from the culture medium 4 hours later, along with control cells that had not been irradiated. Samples were washed twice with PBS buffer and the cells harvested in preparation for RNA extraction.
2) Irradiation
Cells were irradiated with a medical linear accelerator using 0.5, 1, 2 and 5 Gy front and back two-gate irradiation at 0 and 180 degrees. For sufficient irradiation of HeLa cells by the posterior scattered ray, 10 cm thick tissue-equivalent substances covered the upper and bottom surfaces of the culture vessel.
3) RNA extraction
The cells were harvested, as described above, and total RNA was isolated using an RNAgent kit (Promega, Madison, WI). The mRNA was purified with an Oligotex Kit (Qiagen, Valencia, CA), the total RNA concentrations quantified by spectrophotometry at 260 nm and the presence of 28 S and 18 S bands confirmed by agarose gel electrophoresis.
4) Suppression subtraction hybridization (SSH)
PCR-applied SSH was performed using a cDNA subtraction kit (CLONTECH, Palo Alto, CA). Briefly, SSH was performed by synthesis of cDNA from the mRNA of testers (irradiated with 2 Gy x-ray) and drivers (control cells, without irradiation). The cDNA was then treated with restriction enzymes to generate short fragments. Subsequently, adaptor molecules were attached only to the testers, and the primer binding site was obtained by PCR. Finally, a library of differentially expressed genes only present in the testers was obtained by performing two cycles of PCR and cloning of the PCR products.
(1) Driver preparation
Driver cDNA was prepared by applying 1 ng oligonucleotide primer to 2 µg poly(A)+ RNA. According to the CLONTECH s protocol, blunt-ended cDNA was produced by T4 DNA polymerase. The resulting cDNA pellet was resuspended in distilled water, and the 50 µl reaction mixture digested with Rsa I at 37℃ for 3 hours. The digested DNA was phenol-extracted, precipitated with ethanol and dissolved in 7 µl distilled water.
(2) Tester preparation
Various adaptors, which had been digested in the same way as the drivers, were attached to the testers. To 2 µl diluted tester cDNA, 2 µl adaptor 1 or adaptor 2 (10 M), T4 DNA ligase (0.5 units) and ligation buffer were added and the final volume adjusted to 10 µl. After incubation at 16℃ overnight, the adaptor was ligated and 0.2 M EDTA added. Tester samples were heated at 70℃ for 5 minutes to inactivate the ligase, and stored at -20℃.
(3) Subtractive hybridization
To 2 µl cDNA, ligated with 2 adaptor 1 or adaptor 2, were added 2 µl cDNA and 1 µl 4x hybridization buffer. Following the addition of mineral oil, the mixed sample was denatured at 98℃ for 1.5 minutes, and annealed at 68℃ for 10 h. After completion of the first hybridization reaction, two hybridization samples were mixed and 1.5 µl freshly heat-denatured driver added. The samples were rehybridized at 68℃ for 10 h. After hybridization, 200 µl dilution buffer was added; the mixture was heated at 72℃ for 7 min and then stored at -20℃.
(4) PCR amplification
After the hybridization was completed, the unbound subtraction product was amplified twice by PCR. The first PCR was performed using 1 µl subtracted cDNA, 1 µl P1 PCR primer (5 M), 1 µl P2 PCR primer (5 M) and 22 µl PCR master mixture, prepared using an Advantage cDNA PCR Core Kit (CLONTECH). The reaction conditions were 30 heating cycles of 75℃ for 7 min, 91℃ for 30 sec, 68℃ for 30 sec and 72℃ for 2.5 min, and a final extension polymerization at 68℃ for 7 minutes. The amplification products were diluted 10x with distilled water. The second PCR was performed using 1 µl of the diluted PCR product as the template. This PCR solution was identical to that of the first, except for the substitution of the nested PCR primers PN1 and PN2 instead of the P1 and P2 primers. The reaction conditions involved 17 cycles of heating at 91℃ for 30 sec, 60℃ for 30 sec and 72℃ for 2.5 min. The products of the second PCR were analyzed using 2% agarose gel electrophoresis.
(5) PCR-select differential screening
cDNA derived from control, non-irradiated HeLa cells was used as the driver. Forward-subtracted cDNA was prepared using cDNA of irradiated HeLa cells as the tester. Reverse-subtracted cDNA was prepared using cDNA of non-irradiated HeLa cells as the tester.
The cDNA obtained by PCR amplification, as described in 4-4, was cloned into a TA vector, and transformants selected via a gal/IPTG color assay. Agar plates of LB culture medium, containing 40 µl X-gal (20 mg/mL in dimethylformaldehyde), 4 µl IPTG (200 mg/mL) and ampicillin (100 mg/mL), were inoculated and incubated at 37℃. Several hundred white colonies (color due to mutation of the lacZ gene caused by the insertion of foreign DNA) were identified. Two sets of 96 clones, selected arbitrarily from the forward-subtracted library and reverse-subtracted library, were incubated at 37℃, with shaking for 4 hours in LB medium containing 50 µg/mL ampicillin (Sigma, St. Louis, MO). Plasmid DNA was purified from a 1 µl sample using a Wizard Plus miniprep kit (Promega). PCR (using M13 reverse and forward primers for the TA vector) and 1% agarose gel electrophoresis confirmed that the cDNA had been inserted into the vector. PCR-selected differential screening experiments were performed on 88 arbitrarily chosen transformants for the characterization of some of the genes that were differentially expressed in response to radiation. The PCR products, 88 forward-subtracted and 88 reverse-subtracted clones, β-actin and GAPDH products were denatured in the same amount of 0.6 M NaOH, with 2 µl transferred to a nylon membrane, producing two identical dot blots with 96 dots for each sample. Subsequently, the membranes were hybridized at 68℃ with a forward-subtracted cDNA probe-32P and a reverse-subtracted cDNA probe (reverse Northern blotting). The membranes were rinsed with 2×SSC/0.5% SDS solution and 0.2×SSC/0.5% SDS solution and exposed to x-ray film; the patterns of positive/negative blots were analyzed.
5) DNA sequence analysis
After selection through PCR-select differential screening, the sequence DNA of potentially significant forward-subtracted and reverse-subtracted clones were sought. Transformants were incubated with shaking; their plasmid DNA purified and PCR performed using M13 reverse and forward primers. The PCR products were purified by 1.0% agarose gel electrophoresis, and the DNA sequence determined manually by a chain termination reaction or using an automatic system located at the Biomedical Research Center.
6) Real-time quantitative RT-PCR
Real-time PCR was performed using a LightCycler-DNA Master CYBR Green I Kit (Roche, Mannheim, Germany). The amplification reaction was performed using an iCycler (Bio-Rad, Munchen, Germany).
(1) RNA isolation and cDNA preparation
Total RNA of the driver and the tester were isolated using the RNagent kit (Promega). The cDNA was synthesized as previously described (11).
(2) Primer design and cloning of the target
Specific primers were designed for the target gene using the Primer 3 software program and manufactured by Bioneer (Korea). PCR products were separated, isolated and cloned using the pGEM T easy vector system 1 (Promega) and E. coli JM109 (Promega). After incubation with shaking at 37℃, the plasmid DNA was isolated and used to establish a standard curve.
(3) Real-time PCR
SYBR green detection: The iCYCLER IQ system (Bio-Rad) was used for quantitative PCR involving real-time measurement of the fluorescence of CYBR Green dye. The PCR reaction solution was composed of 2 µl DNA Master 10X CYBR green I (Roche: Taq DNA polymerase, reaction buffer, dNTP mix, SYBR green I dye, 10 mM MgCl2), 2.4 µl 25 mM MgCl2, 2 µl cDNA template and 2 µl primer mix (forward: GAGGTACAGCTCGGAACAGC; reverse: TTAGGTGGCTGCCTCAGTTT), along with 11.6 µl PCR-grade dH20, with the final volume adjusted to 20 µl. The reaction conditions were 40 cycles of 94℃ for 30 sec, 60℃ for 60 sec and 72℃ for 30 sec. After completion of the last cycle of PCR, the temperature was increased from 55℃ to 95℃, and the melting temperature measured at 0.5℃ intervals.
(4) Construction of standard curve
The cloned gene to be quantified (RTP801) was serially diluted 10X, CYBY Green I dye added and the PCR amplification performed, as described above. To increase the reliability, duplicate samples were prepared. In the construction of the standard curve, the concentrations within the standard curve obtained in many previous experiments were taken into account, and prepared a curve constructed in the range of ~10-3 pmol. After completion of the amplification reaction, the CT value graph was prepared using the software provided by the manufacturer.
(5) Validation of specific PCR products using melting curve and quantification of RTP801 transcripts
The PCR product was heated from 55℃ to 94℃ and fluorescence readings taken by a computer at 0.5 sec intervals. A curve was obtained describing the fluorescence at various temperatures, and the temperature at which the fluorescence rapidly decreased (Tm) was determined. Observation of the number of Tm obtained provided information about the specificity of the PCR. The CT value of samples was compared with the standard curve, and the mRNA concentrations of the samples quantified.
RESULTS
1) Differentially expressed genes
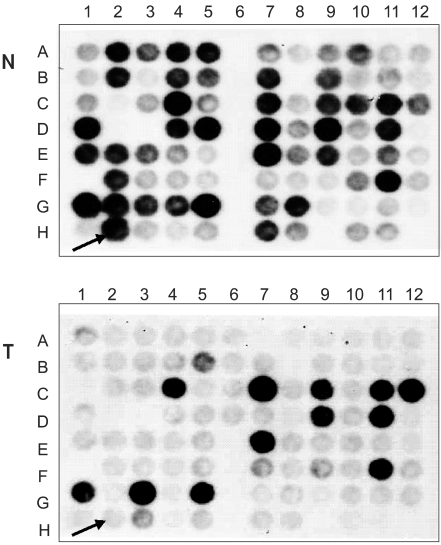
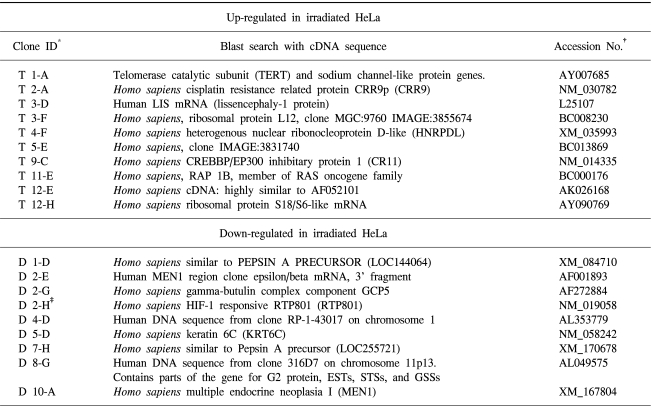
To characterize genes expressed differentially in response to radiation, suppression subtractive hybridization (SSH) was performed on HeLa cells derived from uterine cervical cancer cells. Thirty candidate transformants were selected by reverse Northern blotting. One clone that showed 99% homology with HIF-responsive RTP801 was obtained (Fig. 1).
2) Construction of standard curve
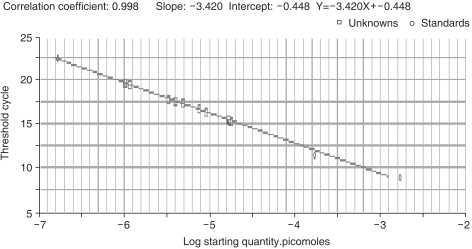
To evaluate the CT value of a specific gene in a gene pool, a CT value standard, exhibiting precise concentrations, needs to be established. Here, the standard curve was established by cloning RTP801 into a pGEM T Easy vector, with subsequent PCR amplification using 10-fold serial dilutions as templates (Fig. 2).
3) Confirmation of PCR products
Double-stranded DNA (dsDNA) is detected through intercalation of UV- or fluorescent-responsive dyes (including CYBR Green dye). Thus, to evaluate the specificity of PCR products, the melting curves were analyzed. The PCR products were heated from 55℃ to 94℃ and the fluorescence measured at 0.5 sec intervals. When the temperature reached the specific melting temperature of a PCR product, the dsDNA separated to ssDNA, the dye intercalated within the double strand was detached, and thus, the fluorescence rapidly decreased. Assessments of these melting curves enable analyses of the specificity of PCRs. The products formed a single band in the RTP801 PCR, which showed that the fluorescent value monitored during the PCR amplification was exclusively from RTP801. The melting temperature was ~89℃ (Fig. 3).
4) Quantification of RTP801 mRNA concentrations by real-time PCR
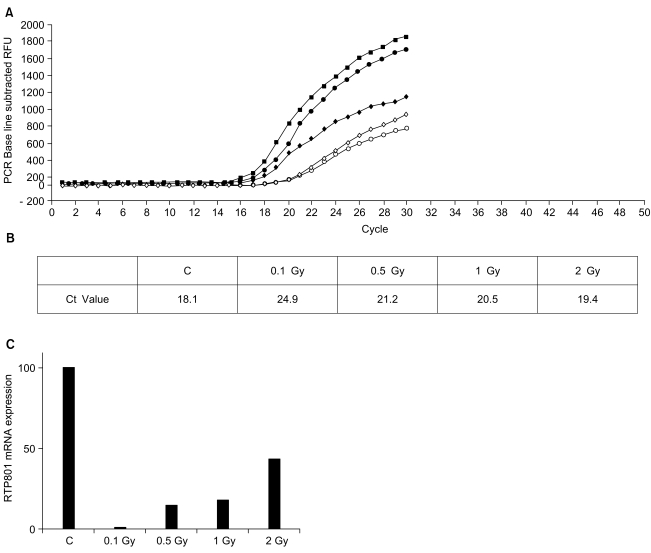
The cDNA was prepared from 5 samples: HeLa cells without irradiation, and HeLa cells irradiated with 0.1, 0.5, 1 or 2 Gy. During the reaction, all programs, including the setting and PCR conditions, were installed in a computer connected to iCycler. To each 2 µl of cDNA, MgCl2 and SYBR Green I were added, according to the manufacturer s instructions, and mixed with the RTP801 primer. During the amplification reaction the real-time reaction product was monitored (Fig. 4A). The CT value representing the threshold cycle was 18.1 in HeLA cells without irradiation, and 24.9, 21.2, 20.5 and 19.4 in HeLa cells irradiated with 0.1, 0.5, 1 and 2 Gy, respectively (Fig. 4B). The values were converted to concentrations by applying the standard curve. The expression of RTP801 mRNA was significantly reduced in irradiated cells compared to the control cells (Fig. 4C).
DISCUSSION
To detect differentially expressed genes in HeLa cells derived from uterine cervical cancer, in response to irradiation, a subtracted cDNA library was prepared by a subtractive suppression hybridization method. Arbitrary selection of 88 clones, and their screening by reverse Northern blotting, enabled the identification of ~30 candidate genes (Table 1). Nucleic acid sequence analyses identified an RTP801 gene, which is a gene of the HIF-responsive gene family. Oxygen is critical for the survival of living organisms, and its intracellular concentrations are tightly controlled. If intracellular oxygen drop below the required level (hypoxia), oxygen-sensing machinery activates a regulatory factor of apoptosis, hypoxia-inducible factor 1 (HIF-1). This, in turn, results in the activation of genes involved in the compensatory mechanisms for cell survival under hypoxic conditions (12). Among HIF-1 target genes is a group of genes that facilitates the delivery of O2 to oxygen-deprived tissues; this group includes erythropoietin, hemeoxygenase 1, vascular endothelial growth factor and inducible nitric oxide synthase (13~16). Another group of genes compensates for the inhibition of oxidative phosphorylation under oxygen-deprived conditions; examples of these include regulatory genes of glycolytic enzymes, such as acetate dehydrogenase (LDH) and phosphoglyceromutase, and the regulatory genes of glucose transporters, such as Glut1 (17~19).
In contrast to reports indicating that HIF-1 target genes are related to adaptive response, Carmeliet et al. reported that the rates of apoptosis increase as the expression of HIF-1 increases, and that the removal of the HIF-1 component HIF-1α suppressed hypoxia-induced apoptosis (20). HIF-1α has been reported to destroy neuronal cells under hypoxic conditions during strokes (21). Recently, the pro-apoptotic gene, Nip3, has been reported to be dependent on HIF-1 (22). In addition, Elena et al. reported that RTP 801, which was significantly reduced in HeLa cell in response to irradiation in this work, was a new HIF-1 response gene (12). In the same article, the low levels at which RTP801 was expressed in various human tissues was described and hypoxia was reported to significantly induce the expression of mRNA in neuronal PC12 cells. Under these circumstances, the increased expression was related to apoptosis. In the present study, the RTP801 mRNA expression was assessed by real-time quantitative PCR. Relative to the non-irradiated control cells, the expression of RTP801 mRNA was reduced by at least 2x in all cells irradiated with 0.1, 0.5, 1 or 2 Gy.
It is generally believed that tumor hypoxia decreases the efficacy of radiation therapy. It has been reported that hypoxia inducible factor (HIF)-1 is the master transcriptional activator of genes regulated by oxygen concentration, and its expression is elevated in most tumor cells (23,24). It follows that the presence of HIF-1 suggests tumor therapy resistance. Thus, our data, in which radiation reduced the RTP801 expression, suggests decreased resistance to radiation. The expression was reduced most significantly in cells irradiated with 0.1 Gy (around 1/10 expression levels observed in control cells). As the radiation increased, the rate of decrease in the expression of RTP801 attenuated (from 0.5 Gy 1/5, from 1 Gy 1/5, 2 Gy by 1/2), which was not easy to explain.
Real-time quantitative PCR methods were used to specifically assess the mRNA concentrations of differentially expressed genes. Conventional PCR has generally been considered inadequate for quantification, as the product is analyzed after the completion of a designated cycle, regardless of the absolute level of marker genes prior to amplification. The amplification levels can vary significantly due to slight differences in materials. In contrast, real-time PCR analyzes the product during the reaction, particularly in the early stages, when amplification is occurring rapidly, thus avoiding the inconsistencies among samples that might arise through the differential exhaustion of materials.
Methods for the quantitative assessment of marker genes have been developed since the late 1990s. Methods involving analyses of SNPs and assessments of splice variants, as well as others, have been used as experimental tools (25). In real-time PCR, a critical parameter is the CT value, that is, the amplification cycle of the fluorescence generated by the separation of probes reaching a designated value above that of the standard. The quantification of genes of unknown concentration is achieved by comparisons with the standard curve. For preparation of the standard curve, the genes to be amplified must be cloned into vectors, and serial 10-fold dilutions of their plasmid DNA amplified by PCR. The initial gene concentration and CT value can then be compared. Herein, triplicate reactions for each sample were prepared, thus increasing the reproducibility of the experiments. This enabled us to obtain a highly reliable correlation coefficient of 0.998 for our standard curve. Real-time PCR detects the fluorescent signals that may be generated through the attachment of fluorescent material to a specific probe complementary to the target gene. Instead, SYBR Green dye, which is a fluorescent material that intercalates non-specifically into dsDNA, was used in our series of experiments. There was a need to consequently evaluate the specificity of the reaction after the PCR amplification, using a melting curve, in this series of experiments. Melting curves are a means of evaluating the melting temperature of dsDNA. The fluorescent values of SYBR Green dye and other DNA-binding dyes are highest when two DNA strands are annealed; when temperatures are greater than Tm, the strands separate and the fluorescent value suddenly decreases. The rate of change may be obtained by plotting -F/dT against temperature. The peak is formed at the largest rate of change, which is the Tm of the dsDNA PCR product. Each DNA has a specific Tm; and therefore, the number of peaks represents the number of PCR products. Only one peak was obtained in this work (Fig. 3), which indicates that the major fluorescence was that of the desired product, with a Tm of 89℃.
CONCLUSION
We have shown for the first time that the mRNA expression levels of the RTP801 gene significantly decrease in irradiated HeLa cells. This may be critical for understanding the expression of genes in cells in response to irradiation. However, further investigations are required in order to understand the biological implications of our result, and their relation to the resistance or sensitivity of cells to radiation.