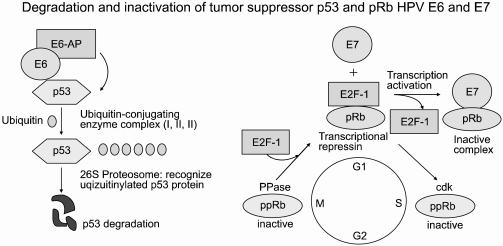
INTRODUCTION
Human papillomavirus (HPV) is the most prevalent sexually transmitted infection in the world, occurring at some point in up to 75% of sexually active women. Although HPV infection is widespread, few people even know they are infected as the symptoms are seldom noticeable. It is even less well known that nearly all cervical cancers (99.7%) are directly linked to previous infection with one or more of the oncogenic types of HPV (1). It is estimated that for every 1 million women infected, 10% (about 100,000) will develop precancerous changes in their cervical tissue. Of these, about 8% will develop early cancer limited to the whole epithelial layer of the uterine cervix (carcinoma in situ; CIS), and a few will develop invasive cancer unless the precancerous lesions are detected and treated, with such cases having been found to carry the oncogenic HPVs (e.g., types 16 and 18) that cause cervical cancer. HPVs have circular, double-stranded DNA genomes that are approximately 8 kb in size and encode eight genes, of which E6 and E7 have transforming properties. These proteins have pleiotropic functions, such as transmembrane signaling, regulation of the cell cycle, transformation of established cell lines, immortalization of primary cell line and regulation of chromosomal stability. The viral E6 and E7 oncoproteins are necessary for malignant conversion. The abilities of high-risk HPV E6 and E7 proteins to associate with the tumor suppressors p53 and pRB, respectively, have been suggested as a mechanism by which these viral proteins induce tumors (Fig. 1). The E6 protein consists of 158 amino acid residues and contains two zinc-finger binding motifs (2). The E6 protein is thought to promote cell proliferation by stimulating degradation of the tumor suppressor p53 protein via the formation of a trimeric complex comprising E6, p53 and the cellular ubiquitination enzyme E6-AP. E6-stimulated degradation interferes with such biological functions of p53; thus perturbing the control of cell cycle progression, leading finally to increased tumor cell growth (3). Although it is commonly accepted that the ability of high-risk type HPV E6 to target p53 for degradation contributes to virus-induced cellular transformation, it is also clear that the E6 protein has oncogenic activities that are independent of p53.
The E7 proteins encoded by the high-risk type HPVs, such as HPV 16 and HPV 18, bind Rb with a much higher affinity compared to those encoded by the low-risk type HPVs, such as HPV 6 and HPV 11. E7 binds to a region of the Rb protein commonly referred to as the 'pocket domains' (4). The 'pocket domain' sequences of Rb are essential for its tumor suppressor function, with many naturally occurring loss-of-function mutations of Rb appearing to cluster in these 'pocket domains'. One of the major biochemical functions of Rb is to bind E2F-family transcription factors and repress the expressions of replication enzyme genes (5). The ability to repress the expressions of replication enzyme genes correlates with the tumor suppression function of Rb. E7 disrupts the interaction between Rb and E2F, resulting in the release of E2F factors in their transcriptionally active forms (6). This E7-mediated conversion of E2Fs to their activator forms stimulates replication and cell-division, which is consistent with the observation that keratinocytes constitutively expressing E7 remain replication- competent, even after differentiation (7). Therefore, complex formation between the products of oncogenes and tumor suppressor genes is believed to be important in the cellular transformation that leads to the disruption of the normal physiological functions of the specific tumor suppressor gene products.
Cervical cancer-specific biomarkers, as yet, have not been well characterized. The diagnosis of cervical cancer is based on histological examinations, with the prognosis assessed based on the clinical features, such as the stage of the disease at diagnosis. Host proteins associated with HPV E6 and E7 oncoproteins should contain fundamental information about the development and progression of HPV-associated cervical carcinogenesis. Utilization of this information for discovering biomarkers, and/or therapeutic targets could be possible if a tool can be developed to rapidly analyze and display changes in protein expression under different conditions for neoplastic cell subpopulations. Therefore, the search for proteins associated with a given cancer is a major challenge.
HOST PROTEINS ASSOCIATED WITH HPV E6 ONCOPROTEIN
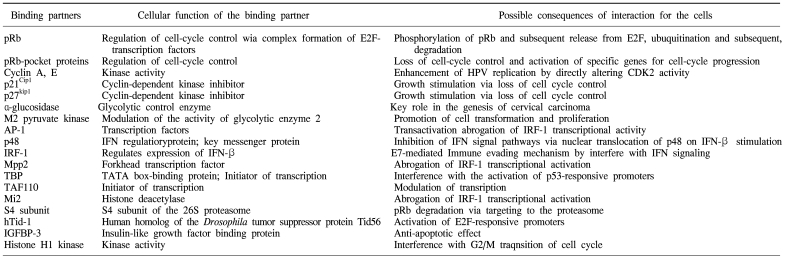
The E6 oncoproteins of high risk HPV interfere with the function of the cellular tumor-suppressor protein p53 through the induction of increased proteasome-dependent p53 degradation. High risk HPV E6 proteins target the cellular E3 ubiquitin ligase E6AP to p53, resulting in the transfer of ubiquitin peptides from E6AP to p53, which marks p53 for degradation by the 26S proteasome. Low risk and cutaneous epithelia-infecting HPV E6 proteins are unable to target the cellular p53 protein for degradation through the proteasome pathway. Although E6-induced loss of p53 is an important element of E6-induced cellular transformation, recent studies have identified a number of additional cellular targets of E6 that may also play an important role. These included the following (8, 9): proteins involved in the regulation of transcription and DNA replication, such as p300/CBP, Gps2, IRF-3, hMcm7, E6TP1 and ADA3; proteins, involved in apoptosis and immune evasion, such as Bak, TNF receptor 1 (TNF R1), FADD and c-Myc,; proteins involved with epithelial organization and differentiation, such as paxillin, E6BP/ERC-55, zyxin and fibulin-1; proteins involved in cell-cell adhesion, polarity and proliferation control, which contain a PDZ-binding motif, such as hDLG, hScrib, PKN, MAGI-1, MAGI-2, MAGI-3 or MUPP1; and proteins involved in DNA repair, such as XRCC1 and 6-O-methylguanine-DNA methyltransferase (MGMT) (Table 1).
In this article, we described the global pattern of protein expression in human adenocarcinoma cell lines stably expressing HPV E6 gene, compared with those of control cells, using a proteomics analysis. An interesting study in comparative proteomics showed the expression of proteins in response to transfection by E6. The prominent function of HPV E6 is the proteolytic inactivation of certain pro-apoptotic factors, such as p53, Bak or Bax, through the ubiquitin-proteasome pathway (10, 11). The Bcl-2 family of proteins is a critical death regulator of mitochondrial integrity and of mitochondria-initiated apoptosis. Bak plays a major role in the apoptosis of several types of cells. The findings of increased levels of Bcl-2 and decreased Bak expression indicates a change in the balance of anti-apoptotic and pro-apoptotic molecules, with a shift in favor of the former. Although Bcl-2 appears to bind poorly to Bak, the over-expression of Bcl-2 is shown to suppress the death function of Bak (12). Therefore, the imbalance between Bcl-2 and Bak is believed to play a role in promoting growth in E6-infected cells. TRAF-interacting protein (I-TRAF) has been shown to be up-regulated by E6. I-TRAF, a novel signal transducer of the TNFR superfamily, inhibits the induction of NF-κB activation mediated by TNFR6, which indirectly interacts with TRAF2 via TRADD. However, I-TRAF was unable to inhibit the activation of NF-κB by IL-1R, which is mediated by TRAF6 (13), suggesting a negative effect of I-TRAF on NF-κB activation was specific to a TRAF2- mediated pathway. E6 may suppress the function of I-TRAF as a negative regulator of NF-κB activation. Cellular damage or stress results from two fundamental cellular responses: apoptosis, a precisely programmed form of cell death, and stress response, or the expression of heat shock protein (HSP), which protects cells and mediates an accelerated recovery following damage. HSPs are multi functional molecular chaperons relating to cellular homeostasis. Primary, HSPs are expressed in response to cellular stresses, which may also include carcinogenesis. E6 induced stronger up-regulation of HSP60, HSP70 and HSP90-β proteins in our experiment. There has been a report that the expressions of HSP40, HSP60 and HSP70, but not that of HSP90, were increased with increasing severity of the cervical lesion, suggestive of stress responses from the progression from an HPV infection to cervical cancer.
Many proteins involved in cell cycle progression are expressed, including E2F5 and CDK5 (cyclin-dependent kinase5). Proteins involved in G1/S progression are decreased, and were thought to be concerned with cyclin-dependent kinase regulation. Transformed cells expressing E6 lose the G1 checkpoint activity at a very early point, presumably due to the degradation of p53. The G2 checkpoint is initially unaffected, but there is increased chromosomal instability in E6-expressing cells over time, which is probably caused by the observed attenuation of the G2 checkpoint function (14). This may be caused by E6 altering the cyclin/cdk complexes, or may relate to the ablation of a p53-regulated G2/M checkpoint. The up- regulated proteins have been studied further for their diagnostic potential.
In our previous work, we looked for p53-independent HPV E6 interacting proteins, using a yeast two-hybrid screening, which resulted in the identification of p73; a homologue protein of p53. The functional inactivation of p73, by both high- and low-risk HPV E6, could play an important role in the malignant transformation and benign condyloma formation of the cervix (15).
Recently, we found a novel interaction between HPV E6 and BARD1 (BRCA1-associated ring domain 1), which was described as a crucial partner of the breast and ovarian tumor suppressor BRCA1 (16). We also demonstrated the existence of BARD1 as a p53 tumor co-suppressor, which inactivated the E6 oncoprotein.
HOST PROTEINS ASSOCIATED WITH HPV E7 ONCOPROTEIN
HPV E7 proteins interact with the so-called 'pRb-associated pocket proteins', including the retinoblastoma protein pRb, which are negative cell-cycle regulators involved in the G1/S and G2/M transitions. The interaction between high-risk E7 and pRb results in enhanced phosphorylation and degradation. pRb destruction leads to the release of E2F family of transcription factors and the subsequent activation of genes promoting cell proliferation. However, the stimulatory effect of E7 upon cell proliferation not only depends on its association with pRb, since E7 targets the function of a plethora of cell cycle regulators, including cyclin A, E and cyclin-dependent kinase inhibitor p21Cip1 and p27kip1 together with the metabolic regulators, acid-α-glucosidase and M2 pyruvate kinase. HPV E7 also interferes with the activity of a variety of cellular transcription factors, such as AP-1, p48, interferon regulatory factor-1 (IRF-1), forkhead transcription factor MPP2, TATA- box binding protein (TBP) and TATA-box binding protein- associated factor (TAF110), as well as with the Mi2 histone deacetylase activity. Also, E7 interacts with the S4 subunit of the 26S proteasome, a human homolog of the Drosophila tumor suppressor protein Tid56 (hTid-1), insulin-like growth factor binding protein (IGFBP-3) and histone H1 kinase (Table 2) (17, 18).
In a proteomic study using E7-transfected C33A cell lines, cytoskeletal proteins, such as actin and myosin V, were down-regulated by E7 (19). Tumor suppressor integrase interactor 1 protein (INI), ING1, and a novel protein, DJ579F20.2.1, (similar to transcriptional repressor CTCF) were suppressed by E7. E7 was also found to down-regulate chaperones, such as protein disulfide isomerase (PDI) A3. INI is a member of the ATP-dependent SWI/SNF chromatin remodeling complex, also known as tumor suppressor hSNF5/INI, which is mutated in sporadic and hereditary malignant rhabdoid tumors (20), causing G0-G1 cell cycle arrest (20). It was recently reported that the SWI/SNF complex plays an important role in p53-regulated cell cycle control (21), suggesting that the E7-induced down-regulation of hSNF5/INI may contribute to oncogenesis. ING1 is also suppressed by E7, and is a candidate tumor suppressor gene, which contributes to the process of malignant transformation and progression (22). ING1 is involved in DNA repair, apoptosis and chromatin remodeling, and is known to share several biological functions with p53, including cell cycle arrest, DNA repair and apoptosis, and is down-regulated in various cancers, such as breast and gastric cancer (22). However, the relationship between ING1 and the HPV oncoprotein remains to be reported. The mechanism by which E7 inhibits the expression of ING1 should also be elucidated. PDI is an enzyme that catalyzes the formation and isomerization of disulfide bonds, which has peptide binding and chaperone activity for cell adhesion, and is also involved in cell-cell interaction and gene expression. This report demonstrates that E7 can evade immune surveillance and resist apoptosis by suppressing or inducing various factors, such as immune- regulatory, cell signaling and cell cycle regulatory factors.
In HaCaT keratinocytes, E7 was found to down-regulate cytoplasmic actin and the leukocyte elastase inhibitor (LEI) (23). Actin plays an essential role in regulating cell structure during reorganization of cells via specific association with tight junctions. Neutrophil elastase is a protease involved in tissue destruction and inflammation, which characterizes numerous diseases, including hereditary emphysema, chronic obstructive pulmonary disease, cystic fibrosis, adult respiratory distress syndrome, ischemic-reperfusion injury and rheumatoid arthritis (24). Thus, elastase has been the object of extensive research in the development of potent inhibitors targeting its destructive and pro-inflammatory actions (24). This data suggests E7 may be involved in the modulation of elastase activity due to the down-regulation of LEI expression. Chaperone-related proteins, such as HSP60 and cytosolic chaperonin-containing T-complex polypeptide 1 (CCT), were up-regulated. HSP60 is known to be highly expressed in various cancers (25), is presumed may interact with a variety of cellular proteins and has a role in cervical carcinogenesis (25). CCT is a molecular chaperone that plays an important role in the folding of proteins in eukaryotic cytosol. Actin, tubulin and several other proteins are known to be folded by CCT. CCT expression is strongly up-regulated during cell growth, especially from the G1/S transition to the early S phase. CCT is expected to play important roles in the growth of cancer cells by assisting the folding of tubulin andother proteins (26). Ku70BP was up-regulated by E7 in 2-DE, with Ku70 and Ku80 essential for DNA double-strand break repair. It has been suggested that Ku70BP amplification and overexpression may be involved in the function of Ku70 in the DNA-PK complex implicated in the maintenance of genome stability and reduction of mutation frequency (27). E7-induced up-regulation of Ku70BP as been assumed to contribute to the functioning of Ku70, which plays roles in double-strand DNA break repair and maintenance of telomeres. Livin inhibitor-of-apoptosis, a novel human inhibitor of apoptosis protein (IAP) family members, termed 'livin', was induced by E7. The mRNA for livin was undetectable in most normal adult tissues, but was present in developmental tissues and several cancer cell lines. Livin was expressed in the nucleus and in a filamentous pattern throughout the cytoplasm. Disruption of livin may provide a strategy for the induction of apoptosis in certain cancer cells (28).
These results suggest that the infection of the E7 oncogene into epithelial cells can evade immune surveillance or resist against apoptosis by inducing or binding to various factors, such as chaperone, cell signaling and cell cycle regulatory factors.
CONCLUSIONS
Cervical cancer is a worldwide public health problem among women, especially in emerging nations. To improve the control of cervical cancer, new adjuvant diagnostic and therapeutic strategies are required. Advances in immunology, genomics and proteomics have accelerated our understanding of the genetic and cellular basis of many cancer types. Cervical cancer is a member of the virus-related neoplasms, with its initiation and promotion associated with persistent infection of oncogenic HPV.
The application of functional genomics and proteomics to query cervical cancer tissue specimens has yielded novel cancer biomarkers, but these require further validation. The integration of molecular and proteomic biotechnology with immunology has also yielded promising findings that may translate into clinically relevant biological assays. The studies of host proteins associated with HPV E6 and E7 oncoproteins continue to display potential future usage in the management of cervical cancer. Subsequent scientific investigations will probably yield new diagnostic and prognostic tools for cervical cancer, provide insights into its underlying biology and contribute to the development of novel management strategies.
Proteomics is now widely accepted as a useful tool in the development of molecular diagnosis and the identification of disease biomarkers in the post-genomic era. Proteomics also promises lower R&D costs, with the opportunities of new revenue streams through the identification of new drug targets in the treatment of various cancers. This review identifies the key technologies that will enable pharmaceutical companies to develop new niche products, improve drug attrition rates, increase the speed of clinical development and target new drug markets.