Introduction
Colorectal cancer (CRC) is the third leading cause of cancer in Korean men and women, and there are an estimated 25,881 new cases each year [1]. An estimated 15% to 20% of CRCs occur in patients with a potentially heritable genetic risk factor [2]. The most common of these genetic alterations is Lynch syndrome (LS), which accounts for approximately 1% to 3% of all CRCs [3–5].
LS is a heritable autosomal dominant genetic condition caused by mutations in several DNA mismatch repair (MMR) genes, MLH1, MSH2, MSH6, PMS2, and EPCAM [6]. Affected individuals have an increased life-long risk of developing multiple cancers, including CRC, endometrial cancer, small bowel cancer, pancreatic cancer, gastric cancer, and genitourinary cancer. Because these cancers occur in relatively young individuals, screening by use of standard recommendations for the general population is ineffective. Methods to identify patients at risk for LS continue to rely on analysis of family pedigrees. However, recent publications have supported universal screening for LS which implements routine tumor testing of all CRCs to identify tumors with microsatellite instability (MSI), a feature of MMR deficient tumors [5,7–10].
Despite efforts to effectively identify, treat, and manage patients with LS, most Korean patients with LS are diagnosed at an advanced stage, thus indicating the need for more effective screening [11]. Most tertiary care centers in Korea now offer routine tumor testing for MSI; however, additional genetic testing is rarely performed. The missed diagnostic opportunities are maybe due to the lack of reports of the yields of these tests for finding LS patients in the Korean population. Thus, our aim was to report the yield of adopting universal screening in comparison with conventional pedigree-based screening for LS in a tertiary center.
Materials and Methods
1. Patients
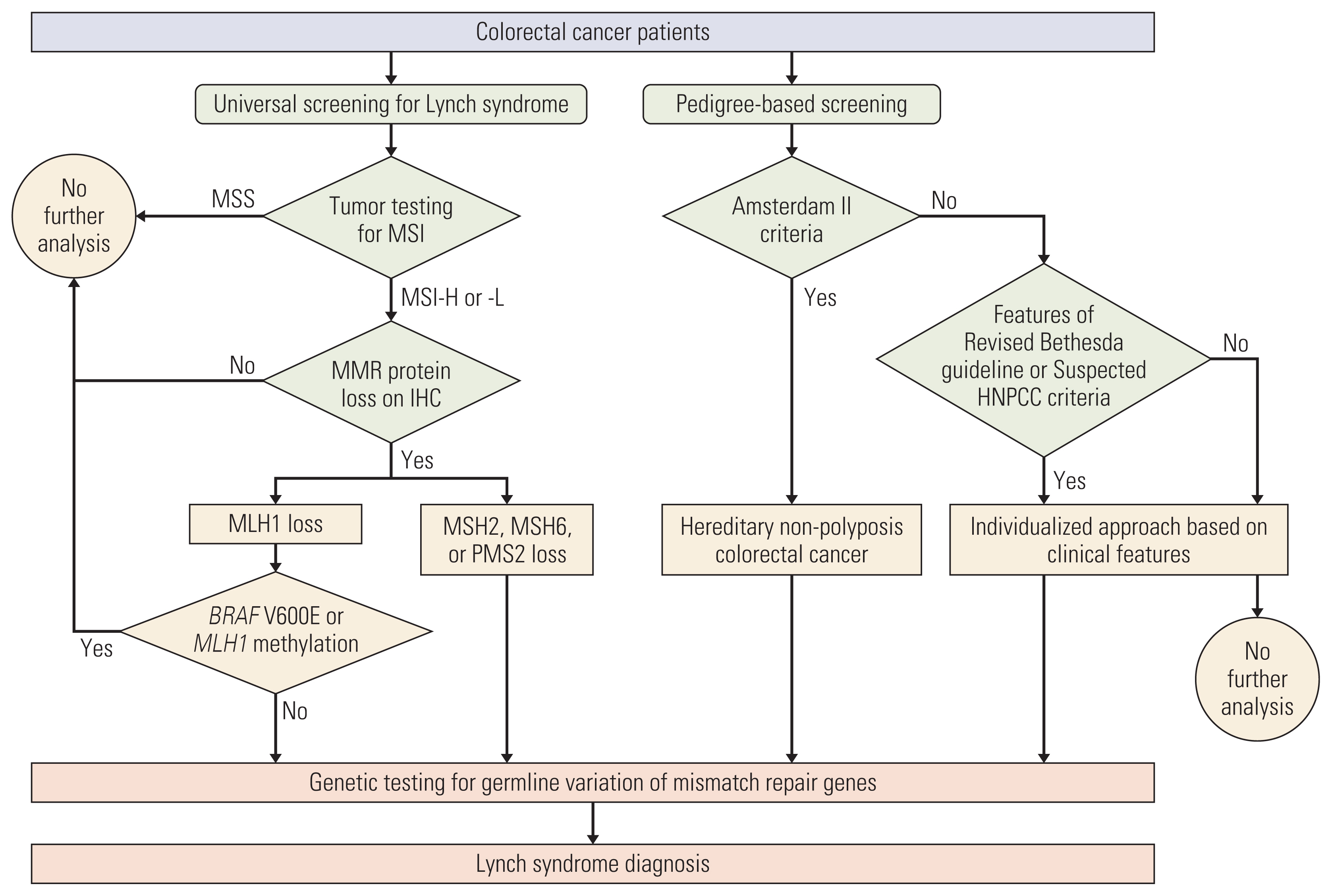
Routine tumor testing for MSI at Seoul National University Bundang Hospital began in 2007. Data from a prospectively collected cohort of CRC patients who received surgery from January 2007 to October 2018 with available data on either tumor testing or family history were retrospectively reviewed. Patients with polyposis syndromes were excluded. When possible, each patient was simultaneously evaluated for hereditary nonpolyposis colorectal cancer (HNPCC) based on family history, along with tumor testing for MSI (Fig. 1). Subsequent immunohistochemistry (IHC) was done for MMR proteins for tumors with high- or low-MSI to narrow down the gene responsible for LS. Patients showing altered patterns for any MMR protein were offered germline genetic testing for the corresponding gene. Germline genetic testing was not further pursued for patients showing pathologic features of somatic tumor mutations related to MLH1 such as BRAF V600E mutations or MLH1 promoter region hypermethylation [12]. Germline genetic testing was done since 2016 when testing was readily available at our center. LS was used to refer to patients with confirmed germline mutations in any one of five mismatch repair genes; HNPCC was used to refer to patients who fulfilled the Amsterdam II criteria, some of whom also had confirmed LS [6,8].
2. Tumor testing for MSI, IHC, and germline genetic testing
The methods for extracting tumor DNA and testing for MSI have been described elsewhere [13,14]. In short, MSI testing was performed using the Bethesda panel. In this test of five microsatellite markers (BAT-25, BAT-26, D2S123, D5S346, and D17D250), instability of two markers indicated high MSI (MSI-H), instability of one marker indicated low MSI (MSI-L), and no signs of instability indicated microsatellite stable (MSS). Tumors showing MSI-H were subjected to IHC testing for four biomarkers (MLH1, MSH2, MSH6, and PMS2) in the pathological sample using antibody cross-linkage (MLH1 [mouse monoclonal primary antibody, prediluted, clone M1, Roche, Basel, Switzerland], MSH2 [mouse monoclonal primary antibody, 1:200, clone G219-1129, Cell Marque, Darmstadt, Germany], MSH6 [mouse monoclonal primary antibody, 1:100, clone 44, Cell Marque], and PMS2 [mouse monoclonal primary antibody, prediluted, clone ERP3947, Roche]). Germline genetic testing was done by direct sequencing (Illumina MiseqDX platform, Illumina, San Diego, CA) with optional addition of multiplex ligation-dependent probe amplification of genomic DNA of the target gene isolated from peripheral blood leukocytes. Variants were then clinically curated by a clinician.
3. Obtaining pedigree and diagnosis of HNPCC
A comprehensive pedigree was obtained from each patient [15]. Each pedigree was obtained by recording information of each member of the biological family who had cancer, the type of cancer, and the age and year of diagnosis. These data were recorded by a well-trained physician’s assistant or research assistant during an interview that lasted about 30 to 60 minutes. To assure the integrity of the pedigree, family members reported as having no history of cancer were double-checked for confirmation. Pedigrees were assessed for high-risk features by using pedigree-based criteria – revised Bethesda guidelines, and suspected HNPCC criteria. Patients fulfilling the Amsterdam II criteria were confirmed as having HNPCC [16,17]. Regardless of negative genetic testing results for LS, all patients and family members with HNPCC were recorded in our registry.
4. Yield analysis
The data recorded for all patients were used to compare the yield of genetic testing for LS following the two approaches: universal screening and pedigree-based screening. For universal screening, the yield of tumor testing for MSI and each subsequent test was recorded and reported in the flow shown in Fig. 1. For the pedigree-based screening, we grouped HNPCC patients fulfilling Amsterdam II criteria first. Then of the remaining patients, we grouped according to having high-risk features in the pedigree where this was defined as pedigrees fulfilling at least one criterion in the Revised Bethesda guidelines [18] or suspected HNPCC criteria [19] (Table 1). We reported the number of patients receiving the test and its yield for each corresponding group. For effectiveness of each approach strategy, we compared the LS patients that were not found using that approach and solely relying on the other approach. We analyzed the reason for this and provided descriptive results.
Results
1. Patients
A total of 5,520 patients received surgery for CRC during the study period. Tumor testing for MSI was performed in 4,701 patients (85.2%) and family histories were obtained from 4,241 patients (76.8%). There was an overlap of 3,422 patients (61.9%) with both tumor testing and pedigree data. We identified 69 patients in the total cohort with hereditary CRC (HNPCC+LS) which included both pedigree-based approach of HNPCC and genetic diagnosis of LS. Respectively, 55 patients were diagnosed with HNPCC and 25 were diagnosed with LS. The overlap was 11 patients (Table 2, patients 1, 2, 8, 9, 12, 15–18, 21, 22) where a prior HNPCC diagnosis and subsequent genetic based LS diagnosis were done. Fourteen patients were diagnosed with LS without prior HNPCC diagnosis and were diagnosed by directly undergoing genetic testing based on suspicious clinical-pathologic features including MSI results.
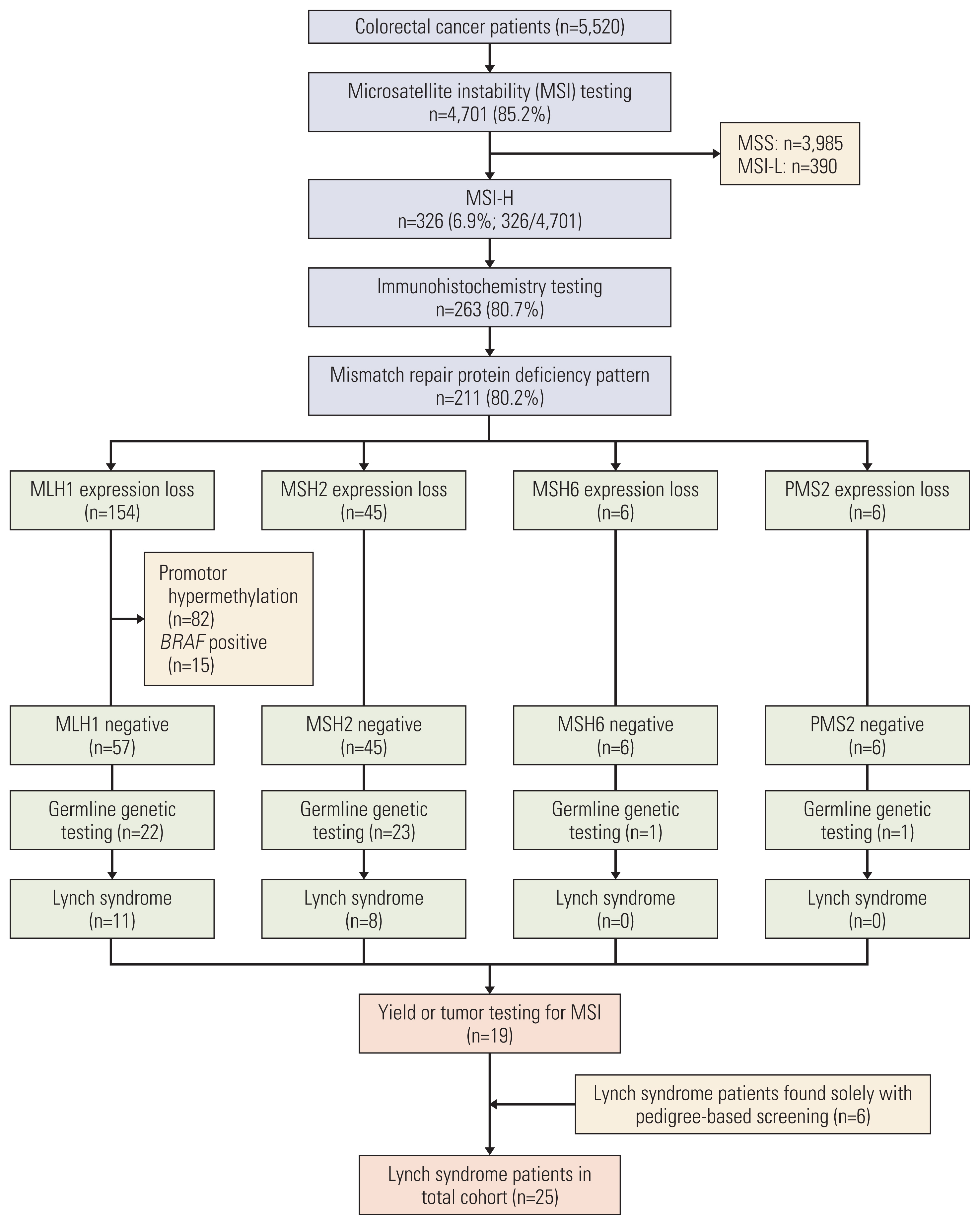
A total of 4,701 of 5,520 study patients (85.2%) received tumor testing for MSI; MSI-H was present in 326 patients (6.9%), MSI-L was present in 390 patients (8.3%), and MSS was present in 3,985 patients (84.8%). Tumors that were too small and could not provide adequate sample for testing (45.5%, 373/819) was the main reason that prevented MSI testing. IHC staining was possible in 263 patients with MSI-H (80.7%, 263/373), and 211 (80.2%, 211/263) had altered protein patterns.
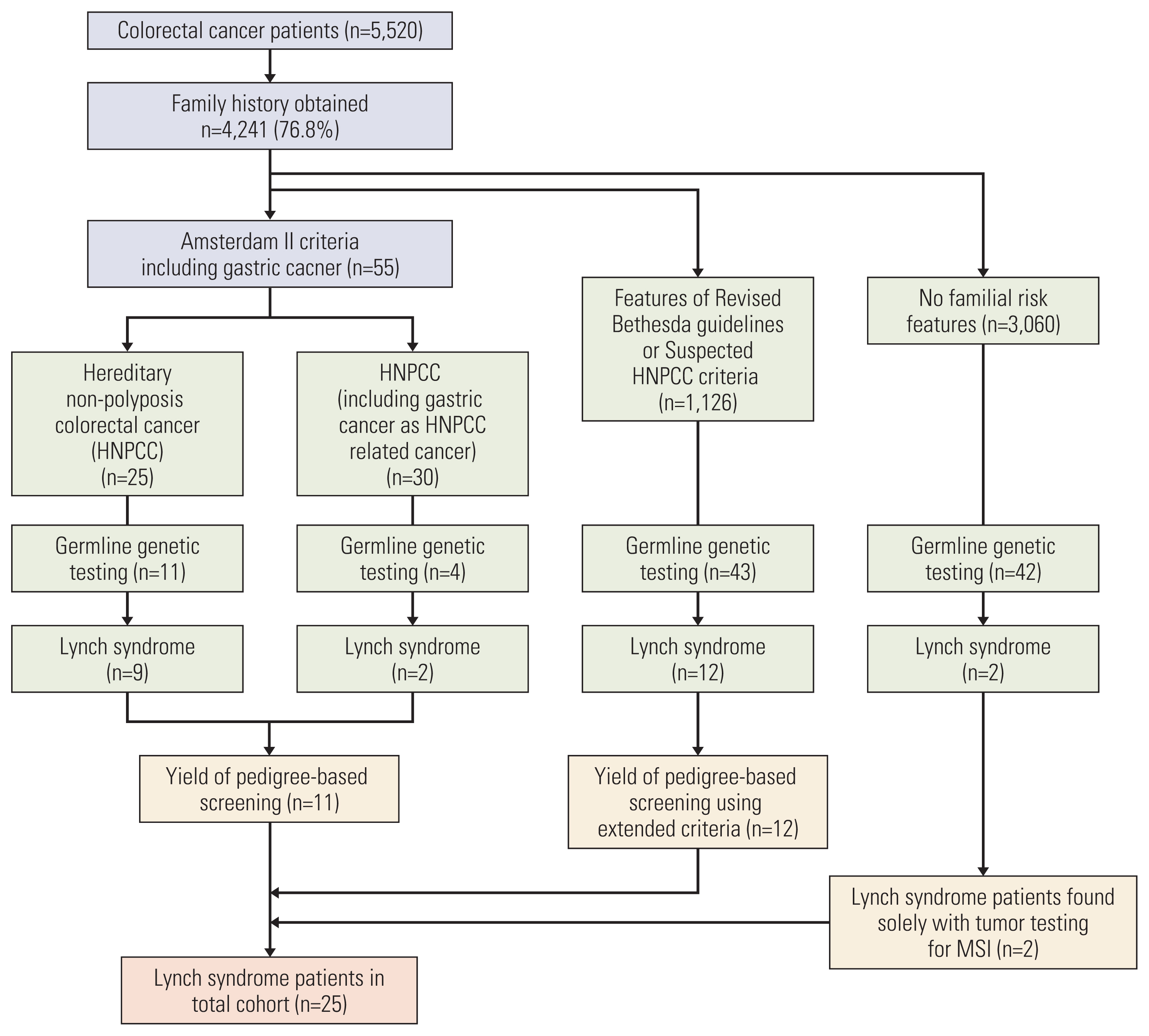
We analyzed family histories in 4,241 of the 5,520 patients (76.8%). Fifty-five of these patients (1.3%) had confirmed HNPCC; 1,126 patients (26.6%) fulfilled at least one criterion of the modified Bethesda guidelines or suspected HNPCC criteria; and 3,060 (72.2%) did not have any familial risk features in the pedigree.
2. Yields of universal screening
Excluding somatic mutations as causes for MLH1 deficiency, there were 57 potential LS cases with MLH1 mutations, 45 with MSH2 mutations, six with MSH6 mutations, and six with PMS2 mutations (Fig. 2). Testing for germline genetic variants was performed in 47 patients (41.2%, 47/114). A positive diagnosis for LS was made in 19 patients (40.4%, 19/47); 11 in MLH1 (50.0%, 11/22), eight in MLH2 (34.7%, 8/23), and none for MSH6 and PMS2 (Fig. 2). This led to an overall yield of 19 patients diagnosed with LS with universal screening.
Universal screening missed LS diagnosis in six patients. Among the six patients, four were HNPCC and two had familial risk features. Among the four patients with HNPCC, one patient (Table 2, patient 15) had a tumor with MSS but had an altered MSH2 pattern in IHC and a familial history of HNPCC-related cancer (father: stomach cancer at 69, sister: CRC at 40, and brother: CRC at 30) indicating the need for genetic testing. Two of the four HNPCC patients (Table 2, patients 1 and 8) had tumors with MSI-H, but insufficient IHC staining prevented a conclusive diagnosis. Both also had a family history of HNPCC-related cancer (patient 1: father, two uncles, and cousin with CRC; patient 8: father and uncle with CRC, uncle diagnosed at age 40). Patient 1 received an initial diagnosis of endometrial cancer, but a Tis CRC was found during further workup. Both tumors were excised simultaneously. The other HNPCC patient (Table 2, patient 21) had a tumor with MSI-H but did not undergo IHC staining. Due to this patient’s young age (23 years) and his mother’s history of CRC, genetic testing was performed.
For the other two patients missed by tumor testing, one patient (Table 2, patient 24) had a suggestive family history (father: stomach cancer in 50s; uncle: lung cancer) and due to his young age (23 years), underwent genetic testing. The other missed patient (Table 2, patient 14) had a tumor with MSS, but two years later received a diagnosis of endometrial cancer. Genetic testing at that time led to a diagnosis of LS.
3. Yields of pedigree-based screening
Due to the high prevalence of gastric cancer in Korea and evidence that gastric cancer is an HNPCC-related cancer [1,16,17], we grouped HNPCC patients based on gastric cancer presence in the family pedigree. Among 55 patients who met the Amsterdam II criteria for HNPCC, 25 had fulfilled this without gastric cancer in their family history and 30 had fulfilled it with inclusion of gastric cancer in their family history (Fig. 3). Germline genetic testing in 11 of 25 HNPCC patients (44.0%) and in four of 30 HNPCC with gastric cancer family history patients (13.3%) led to a positive diagnosis in nine (81.8%, 9/11) and two patients (50%, 2/4), respectively for LS. This led to an overall yield of 11 (73.3%, 11/15) patients diagnosed with LS using pedigree-based screening.
Analysis of 4,186 non-HNPCC patients yielded 14 patients with LS. There were 1,126 non-HNPCC patients fulfilling at least one criterion from either revised Bethesda guidelines or suspected HNPCC criteria. Of these patients, 43 received genetic testing and 12 (27.9%) had confirmed diagnoses of LS. The other 3,060 non-HNPCC patients did not have familial risk features in the pedigree. Of these patients, 42 received genetic testing based on tumor testing for MSI and clinicopathologic features. Two patients were confirmed for LS. One was a 58-year-old male (Table 2, patient 23) and the other was a 75-year-old female (Table 2, patient 19), and they both had no signs of increased risk based on family pedigree data. Both were diagnosed using universal screening for LS.
Discussion
Our combined use of universal screening and pedigree-based screening identified 69 patients with hereditary CRC (HNPCC+LS), accounting for 1.3% of the total cohort of CRC patients. We found that tumor testing was more effective than pedigree-based screening in identification of patients with LS. However, use of either strategy alone led to some missed diagnoses, and in some cases warranted high suspicion from the clinicians. Since 2007, our institute has offered routine tumor testing for MSI for all CRC tumors whenever possible. We also obtained large-scale detailed pedigrees [15] for most of the total CRC cohort. To the best of our knowledge, this is the largest study to report the real-world yield of screening for LS in a cohort of consecutive CRC patients from East Asia.
Despite our high volume, the total proportion of LS patients (0.45%) in our cohort was lower than anticipated. Previous studies estimated that about 1% to 3% of all CRCs are due to LS [3–5]. There may be several reasons for this discrepancy. First, our cohort had a lower proportion of MSI-H tumors (6.9%) and a lower uptake (41%) of genetic testing. Our previous study [13] showed that the frequency of MSI-H tumors in Korean patients (5.5%) with CRC was lower than in Western patients (15%–25%) [3,20]. Also, assuming that all patients at risk based on tumor testing received subsequent genetic testing, we estimate that an additional 34 patients would have diagnoses of LS. This would yield an estimated overall incidence of 1.1%, comparable to that in Western cohorts considering the different proportions of MSI-H tumors. A second reason for the discrepancy may be a surge of sporadic CRCs in the Korean population during recent decades because the rapid economic growth in Korea led to rapid changes in lifestyle that increased the risk of somatic CRCs. Also, the establishment of the National Cancer Screening Program in 2004 facilitated the diagnosis of CRCs, especially for patients over age 50 years, and this may have further reduced the proportion of heritable cancers [21]. Lastly, there may be differences in the mutational profiles of different ethnic groups, and there is only limited curation of data on genetic variants [22,23]. There is therefore a need for further studies of genomic variants in Asian populations and curation of these data.
We identified obvious limitations of pedigree-based screening for identification of LS, in line with previous reports [3,4,19,24]. Previous studies showed that pedigree-based screening missed 5% to 50% of LS patients depending on the screening criteria [25,26]. Our use of the Amsterdam II criteria only identified 44% of LS patients in our cohort, although the relatively low uptake of genetic testing for HNPCC may have factored into this. The main reason for this low uptake is the late initiation of germline genetic testing at our center. We started germline genetic testing since 2016, where many patients had already concluded out-patient follow-up. However, even factoring for the low uptake, a significant proportion of LS patients were found outside this approach including two LS patients who had no prior family history and were diagnosed solely by universal screening.
Universal screening however, also failed to identify LS in six patients. The main cause of this was discrepancy between MSI and IHC results. Extending this to the whole cohort, there were 52 patients with discrepant MSI and IHC results. Staining for IHC may vary in intensity and have different sensitivities and specificities based on the specific antibody [27], so interpretation by experienced pathologists is required. The sensitivity of IHC in predicting a germline mutation increases when more MMR proteins are assayed [28]. However, there can be discrepancies in MSI-H and all MMR proteins. This may be because MMR proteins lose function but not immunogenicity [27,28]. All these LS patients had suspected disease based on family history, so further analysis of such discrepant cases may be able to identify additional LS patients.
We confirmed LS syndrome in 73.3% of HNPCC patients undergoing genetic testing, higher than in a previous Korean cohort [29]. The positive yield from genetic testing was highest for HNPCC patients without gastric cancer in their pedigrees. Two of four HNPCC patients with gastric cancer in their pedigrees had positive diagnoses for LS. Although results may vary with more testing in this patient cohort, our results support the inclusion of gastric cancer as an HNPCC-related cancer, in line with previous studies [16,17].
Based on our results, universal screening for LS and pedigree-based screening each has strengths and weaknesses. Thus, we believe all information should be provided to clinicians when considering genetic testing. Further investigation of our data revealed younger age, or presence of synchronous/metachronous LS-related cancers provided more clinical hints to pursue genetic testing. This information may be useful when used in tandem with genetic prediction models such as the PREMM5 model [30]. Experimental analysis showed all 25 patients diagnosed with LS with the exemption of two patients lacking pedigrees scored over 2.5% (Table 2), which is the recommended threshold to undergo genetic testing as recommended by the PREMM5 model. A strong family history is also an important indication for germline genetic testing because not all hereditary CRCs are LS. Additionally, insufficient small tumor samples may also be an indicator to investigate family history. Despite many large hospitals in Korea offering routine tumor testing for MSI, this information is generally reserved for prognostic implications and prediction of chemotherapy response and there are still many missed diagnoses of LS. Our data may aid practicing surgeons as a basis for genetic counseling and help persuade patients to undergo genetic testing for LS.
Our study had several limitations. First, there was heterogeneity in the diagnostic methods used throughout the 11-year study period. In particular, prior to 2011 IHC testing was not routinely performed for all four MMR proteins in MSI-H tumors, and this may have contributed to an under-diagnosis. Also, a change in health insurance policy in 2016–2017 meant that IHC staining was only covered after confirmation of MSI. During this transition, many tumors were not checked by MSI and IHC, and this also may have contributed to an under-diagnosis. It is thus important to have a dedicated and knowledgeable personnel when implementing screening methods for LS [31]. Third, there was low uptake of genetic testing due to the retrospective nature of the study and delayed initiation of genetic testing. Future prospectively controlled studies may thus result in higher yields for LS diagnosis. Lastly, the generalizability of our findings is limited because all CRC patients were from a single tertiary care center in Korea.
We reported the real-world diagnostic yields of genetic testing for LS in 5,520 Korean patients undergoing surgery for CRC. Hereditary CRC was found present 1.3% of patients and LS was diagnosed in 0.45% of patients. Despite widespread adoption of routine tumor testing for MSI, this is not a fail-safe approach to screen all LS patients. Obtaining a thorough family history in combination with universal screening provides a more comprehensive ‘universal’ screening method for LS.