Introduction
Radiotherapy (RT) is one of the main strategies of cancer treatment, along with chemotherapy and surgery. The main goal of RT is to eradicate cancer cells. Although high dose of radiation can kill large number of cancer cells, they can also cause severe side effects in normal tissues. Thus, in clinical practice, the prescription dose of RT is often limited by the tolerance dose of surrounding normal tissues. To compensate for the reduced RT effect due to an insufficient dose, combined therapy is usually conducted. One such method for cancer therapy is the combination of RT and chemotherapy. However, chemotherapeutic agents not only enhance the radiosensitization of cancer cells but also cause severe systemic side effects that are different from those of RT [1,2]. To achieve maximal tumor control with minimal side effects, the combined treatment including RT and several pharmaceutical agents has been investigated, but further research on novel agents is needed [3].
Alpha-lipoic acid (ALA) is an organosulfur compound derived from octanoic acid, a potent antioxidant that is naturally obtained from humans as well as plants and animals. The effectiveness of ALA in various diseases such as Alzheimer’s disease, cognitive dysfunction, hypertension, diabetes mellitus, and cancer has been demonstrated in several clinical trials [4]. In cancer research, ALA is known to reduce tumor cell viability and proliferation [5]. In addition, several studies have reported that these effects of ALA on cancer are synergistic when combined with chemotherapy [6,7]. Tripathy et al. [8] have recently reported that ALA also enhances the anti-tumor effect of RT on breast cancer cells. Moreover, ALA exerts the opposite effect on normal tissues. The molecule has been proven to reduce the normal tissue damage by decreasing the levels of radiation-induced reactive oxygen species (ROS); therefore, acting as a radioprotector [9,10]. However, the mechanism of the combined ALA and RT treatment remains unclear.
Despite RT being the main strategy for treating cancer, some cancer cells are resistant to radiation-induced cell death, including apoptosis [11]. Therefore, an alternative target to apoptosis is required for the treatment of tumors. Radiation-induced senescence consists of an alternative anti-tumor mechanism in apoptosis-resistant cancer cells [12]. This study aimed to observe the synergistic effect of ALA and RT in terms of cell senescence as well as radiation-induced apoptosis in MDA-MB-231 human breast cancer cells, which are known to be radioresistant.
Materials and Methods
1. Cell culture and treatment
Human metastatic breast cancer cells (MDA-MB-231, Korean Cell Line Bank, Seoul, Korea) were cultured in RPMI-1640 medium with 10% fetal bovine serum and subcultured using 0.05% trypsin-EDTA solution (Gibco BRL, Gaithersburg, MD). The cells were divided into four groups depending on the treatment (control, ALA, RT, and ALA+RT). The ALA+RT cells were incubated with 2 mM ALA for 2, 4, 8, and 24 hours and then irradiated with 10 Gy at room temperature using 6 MV photon beam from a linear accelerator (21EX, Varian, Palo Alto, CA). Treated cells were incubated for 24, 48, and 72 hours and then tested for morphology, apoptotic cell death, mitochondrial ROS, and mitochondrial membrane potential. The experimental schemes are presented in S1 and S2 Figs.
2. Morphological evaluation and viability assay
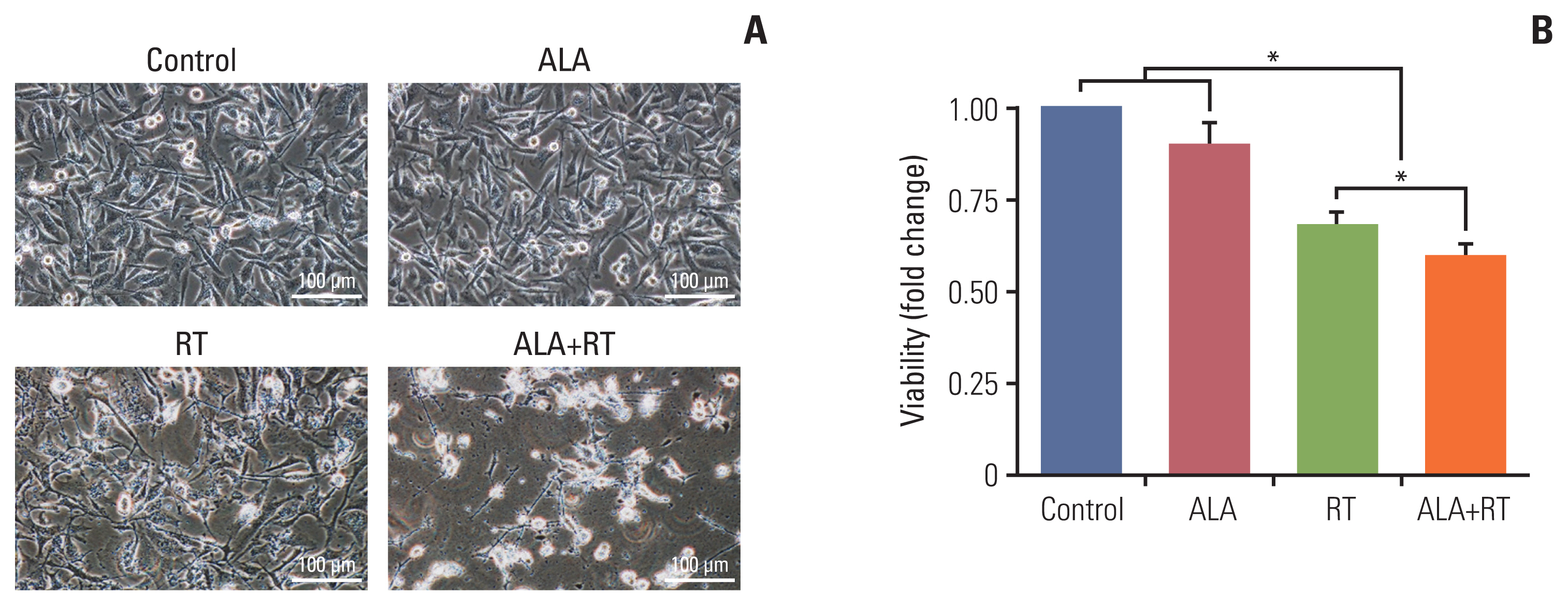
Cells (5×105 cells/dish) were seeded into 60-mm dishes overnight and treated with ALA and/or RT and then analyzed by phase-contrast microscopy for signs of cell death. Signs of cell death were defined as cell shrinkage, rounded cell appearance, and poor cell adhesion. The trypan blue exclusion assay was used to determine cell viability. Viability was represented as a fold change calculated as the ratio of the final test value in each sample to the test value in the control (predefined as “1”). Values were represented as the mean±standard error of the mean (SEM).
3. Flow cytometry for apoptosis detection
Cells were detached, washed twice with phosphate buffered saline (PBS; pH 7.4), centrifuged for 5 minutes at 400 ×g, and resuspended in binding buffer. The cells were then stained using an Annexin V-FITC Apoptosis Detection kit (BD Biosciences, Franklin Lakes, NJ), which was followed by propidium iodide (PI) staining (BD Biosciences). The stained cells were analyzed by flow cytometry (BD LSRFortessa X-20, BD Biosciences).
4. Western blot analysis
Proteins extracts were lysed in RIPA buffer (#89900, Thermo Scientific, Waltham, MA) and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrophoretically transferred to a nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk in PBS-Tween 20 (0.1%, v/v) at 4°C overnight and were incubated with primary antibodies, anti–poly(ADP-ribose) polymerase (PARP; Biomol Research Laboratory, Plymouth Meeting, PA), anti-Bax, anti–Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), anti–caspase-9 (Enzo, Biochem, New York, NY), anti-p53, anti–high mobility group box protein 1 (HMGB1), and anti-phospho-mammalian target of rapamycin (Cell Signaling Technology, Danvers, MA) for 1 hour. Horseradish peroxidase–conjugated anti-rabbit or anti-mouse IgG was used as the secondary antibody. Immunoreactive protein was visualized by the chemiluminescence protocol (Amersham Pharmacia Biotech, Piscataway, NJ).
5. Quantitative real-time PCR
The transcript levels of the cell senescence-related factors were determined by quantitative real-time polymerase chain reaction (qPCR). Cells were resuspended in 1 mL of TRI-zol Reagent (Invitrogen Life Technologies, Carlsbad, CA), and total RNA was extracted. Purified RNA was reverse transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). Quantitative cDNA amplification was performed using a ViiA7 Real-Time System (Applied Biosystems Inc., Foster City, CA) and Power SYBR Green PCR Master Mix (Applied Biosystems). Thermal cycling conditions were as follows: denaturation at 95°C for 3 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds, and annealing and extension at 60°C for 60 seconds. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control for the normalization of the RNA quantity. The relative gene expression level in each sample were quantified using the 2DDCt method. Each primer sequence is as follows.
6. Mitochondrial membrane potential measurement
Mitochondrial membrane potential was measured by staining cells with tetramethylrhodamine methyl ester (TMRM) (Molecular Probes, Eugene, OR). Cells were treated as described in “Cell culture and treatment,” and then incubated with 20 nM TMRM for 20 minutes at 37°C. Images were taken using a Nikon microscope (Nikon, Tokyo, Japan) and the obtained data were analyzed using NIS Elements BR 3.2 (Nikon).
7. Mitochondrial ROS measurement
MitoSOX (Molecular Probes) was used to detect mitochondrial ROS levels in MDA-MB-231 cells. Cells were treated as described in “Cell culture and treatment,” and then incubated with 5 mM MitoSOX for 20 minutes and the obtained data were analyzed by NIS Elements BR 3.2 (Nikon).
8. Cell cycle analysis
MDA-MB-231 cells were cultured and treated with vehicle, ALA, RT, or both ALA and RT. After 72 hours of treatment, cells were harvested, washed, and permeabilized using 70% ethanol at −80°C overnight. Cells were trypsinized and fixed in ice-cold 70% ethanol overnight at 4°C. Cells were washed, stained with PI overnight, and subjected to fluorescence-activated cell sorting by flow cytometry (BD LSRFortessa X-20, BD Biosciences).
9. Statistical analysis
The obtained data were expressed as mean±SEM. Statistical analyses were conducted using the GraphPad Prism 8.0 software (GraphPad Software Inc., La Jolla, CA). The statistical tests of choice were one-way ANOVA with Tukey’s multiple comparison test (for comparison of all groups) with p < 0.05 indicating statistical significance.
Results
1. Effects of ALA on RT-induced changes in cell morphology and proliferation
The representative cell morphologies after each treatment are shown in Fig. 1A. Untreated cells (control) were regularly spread and reached confluence. In other groups (ALA, RT, and ALA+RT), cell number reduction, cell shrinkage accompanied by rounded cell appearance, and poor cell adhesion were observed. However, these changes were more prominent in the ALA+RT group than in the RT and ALA groups. The total numbers of cells were counted (Fig. 1B). Both the RT and ALA+RT groups showed much lower cell numbers after the treatment than the untreated and ALA-treated cells.
2. ALA enhances RT-induced apoptotic cell death
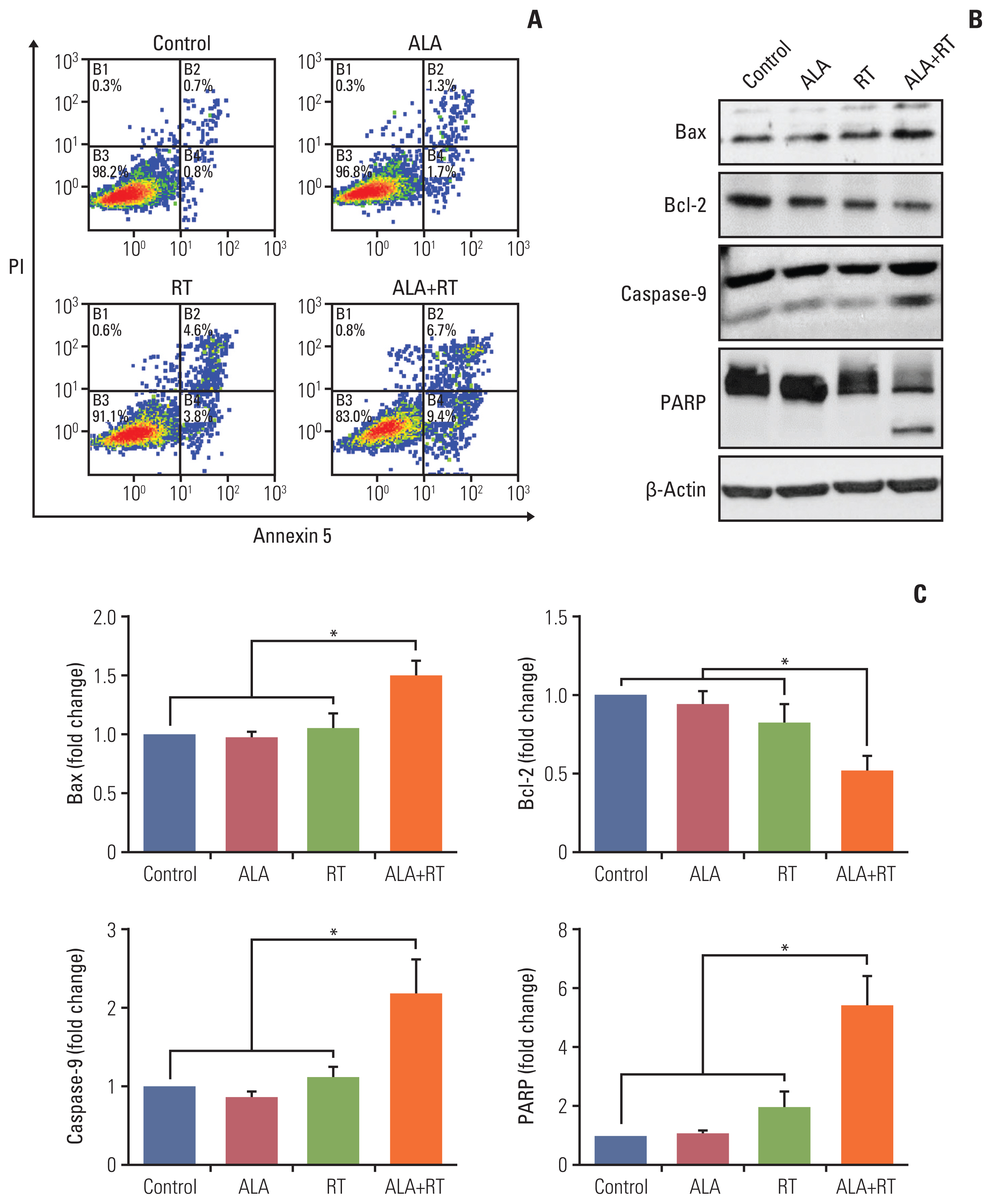
Fig. 2A shows the proportion of viable cells, early apoptotic cells, late apoptotic cells, and necrotic cells in each group. The proportion of early apoptotic cells in the control, ALA, RT, and ALA+RT groups was 0.8%, 1.7%, 3.8%, and 9.4%, respectively, and the proportion of late apoptotic cells was 0.7%, 1.3%, 4.6%, and 6.7%, respectively. The proportion of total apoptotic cells was 1.5%, 3.0%, 8.4%, and 16.1% for each group, respectively, which shows a significantly higher value in the ALA+RT group than in each other group. To additionally evaluate the effect of ALA on apoptotic cell death, apoptosis-related factors were investigated by immunoblotting. The ALA+RT group was characterized by a significantly downregulated expression of Bcl-2 (anti-apoptotic factor) and a significantly upregulated expression of Bax, caspase-9, and PARP (pro-apoptotic factors) compared with other groups (Fig. 2B and C). However, no cleavage of caspase-3 was detected in any of the groups (data not shown).
3. ALA accelerates mitochondrial damage by irradiation
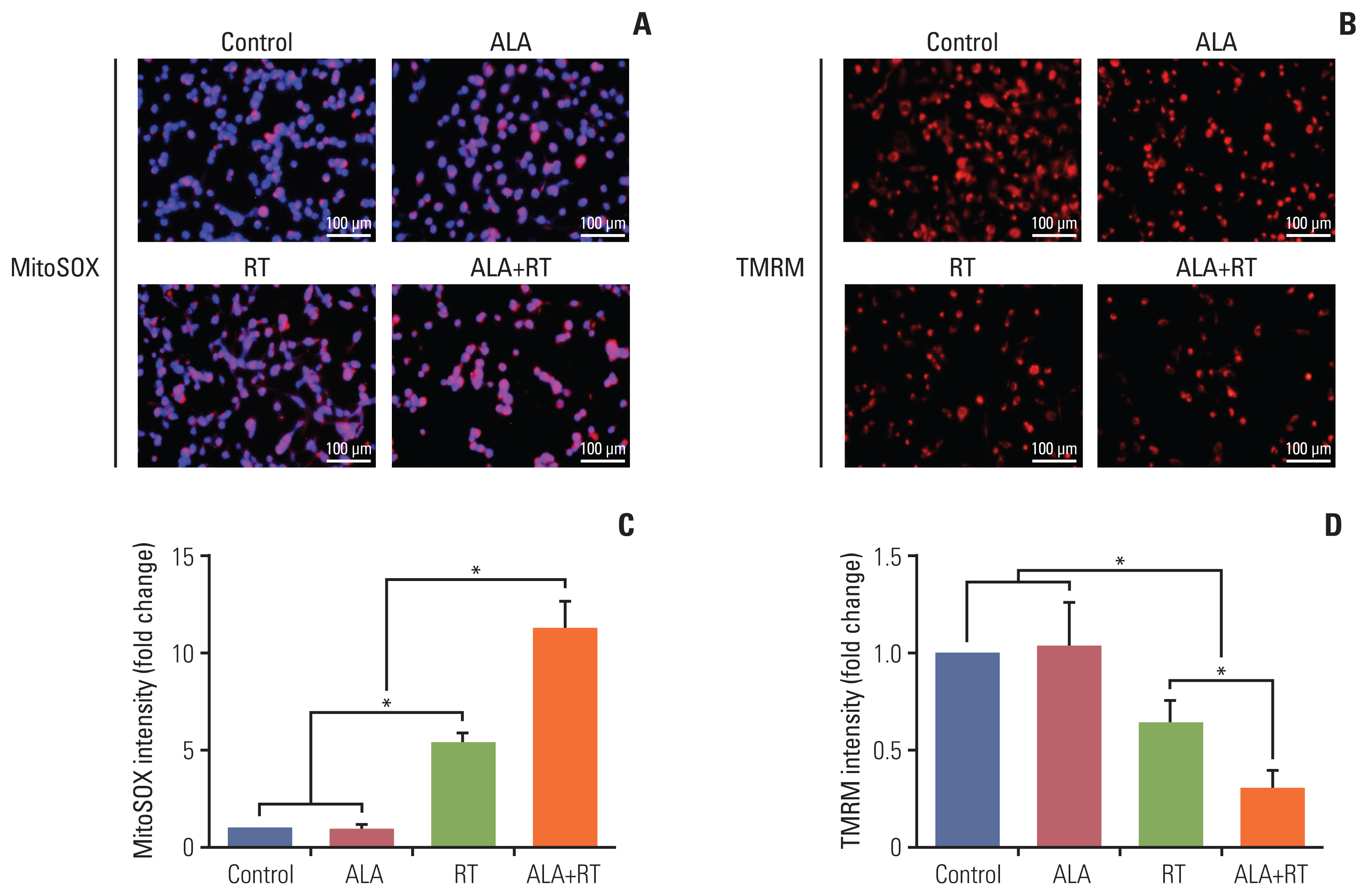
To verify whether ALA is involved in mitochondria-mediated cell death, mitochondrial damage was examined. One of the parameters used to assess the damage was mitochondrial oxidative stress. After each treatment, MDA-MB-231 cells were incubated with the fluorescent probe MitoSOX detecting mitochondria-specific ROS. The density of the positive signals per cell and cell number were significantly increased in the RT group compared to the control as well as the ALA group, and in the ALA+RT group compared with the RT group (Fig. 3A). We also investigated changes in the mitochondrial membrane potential using TMRM dye. The intensity of TMRM-stained cells was significantly decreased in the RT group compared to the control and ALA, and the lowest intensity was observed for the ALA+RT group (Fig. 3B).
4. ALA enhances RT-induced cellular senescence by HMG-B1
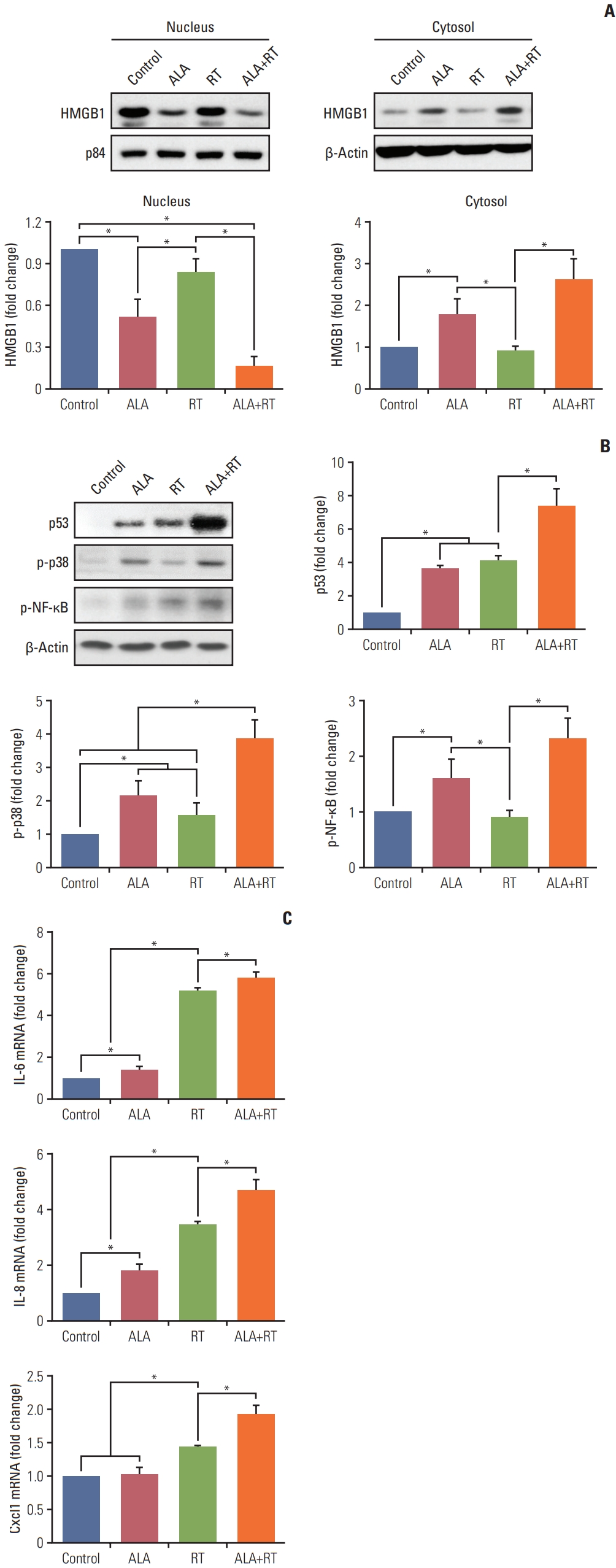
RT is known to induce cellular senescence in tumors [13]. There is also evidence that HMGB1 is indispensable for senescence induction and p53, p38, and nuclear factor κB (NF-κB) activation is associated with radiation-induced senescence [14–16]. As shown in Fig. 4A, cytosolic expression of HMGB1 was increased in the ALA and ALA+RT groups, whereas nuclear expression levels of HMGB1 were decreased in these groups compared to the control. Moreover, expression levels of p53 and activation of p38 mitogen-activated protein kinase (MAPK) and NF-κB were significantly increased in the ALA+RT group compared to the control. The expression levels of IL-6, IL-8, and Cxcl1 (the senescence-associated cytokine and chemokines [17]) were measured by qPCR to additionally examine the effect of ALA on cellular senescence in irradiated cells. Increased levels of the corresponding mRNAs were observed in the ALA+RT group compared to all groups (Fig. 4C).
5. ALA enhances RT-induced cell cycle arrest and expression of cell cycle regulators
Senescence is also defined as permanent cell cycle arrest and inhibits the proliferation of cancer cells [15,18]. The cell cycle was analyzed in all experimental groups and ALA pretreatment showed increased G2/M arrest compared to RT alone (Fig. 5A). In addition, the expression of p21, a cyclin-dependent kinase (Cdk) inhibitor that induces senescence through cell cycle arrest, was significantly increased in the ALA+RT group compared to the other groups (Fig. 5B). The expression of phosphor-Cdk1/2 (upstream factor for p21) was corresponding to the p21 expression pattern.
Discussion
Despite apoptosis being the major cause of RT-induced cell death, some solid tumors exhibit resistance to radiation-induced apoptosis [19,20]. Hence, strategies for triggering the alternative cancer cell death mechanisms might contribute to improved prognosis in cancer patients. Senescence is known to be a tumor-suppressive mechanism that inhibits cancer cell proliferation [15,18]. Ionizing radiation can trigger irreversible cellular senescence by causing DNA damage, oxidative stress, and telomere dysfunction [21].
In the current study, we evaluated the synergistic effect of ALA and RT on cell death and cellular senescence in MDA-MB-231 human breast cancer cells, which are known to be resistant to RT. Our data showed that ALA significantly promotes apoptotic cancer cell death when combined with RT, as reflected by Annexin V staining, expression of apoptosis-related factors, mitochondrial damage, changes in cell morphology, and reduction of cell numbers. In addition, ALA significantly enhances RT-induced cellular senescence through increased HMGB1 expression in the cytosolic fraction. To the best of our knowledge, this is the first report that shows the involvement of ALA in the HMGB1-mediated radiosensitizing effect in cancer cells.
HMGB1 is a histone-like protein that functions as a chromatin-binding factor in the nucleus. On the other hand, in the cytosol, it acts as an extracellular signaling molecule and damage-associated molecular pattern molecule. Moreover, HMGB1 mediates cell damage due to oxidative stress caused by various environmental factors, including RT. It interacts with p53 and enhances its activity [22,23]. In addition, Davalos et al. [16] reported that reduced or elevated HMGB1 expression causes cell senescence and stimulates p53 expression in radiation-induced senescent cells, and loss of nuclear HMGB1, as well as cytosolic HMGB1 secretion, requires p53 activity. Moreover, p53-deficient cells did not secrete or lose nuclear HMGB1 in response to senescence-inducing stimuli. HMGB1-overexpressing senescent cells secreted both HMGB1 and IL-6, whereas HMGB1-depleted senescent cells secreted reduced levels of IL-6 [16]. Our results (Fig. 4) for the ALA+RT group showed increased cytosolic and decreased nuclear HMGB1 expression as well as increased expression of p53, IL-6, and IL-8. These findings are well correlated with previous studies [16,22,23]. In addition, IL-6 levels and NF-κB activity are known to be correlated in the senescent cells, and NF-κb activity was elevated in cells overexpressing HMGB1 [14,16].
Cell senescence is regulated by various factors such as DNA damage, stress, and cell type. It plays an anti-tumor role by forcing cancerous cells into a permanent cell cycle arrest [16,24]. This growth arrest by senescence is established and maintained by the p53 and pRb pathways. The p53-p21waf1/cip1 (p21) pathway and p16INK4a are important for the onset of senescence and the maintenance of senescence, respectively [14]. Cdk inhibitors p21 and p16INK4a cause dephosphorylation of Rb and thereby block cell cycle progression [18]. Chk2 promotes senescence by acting downstream of ATM and thereby inducing p21 [19,20]. Senescent cells increase the expression and secretion of various cytokines, chemokines, matrix metallopeptidases, and other proteins. p38 MAPK is activated during the radiation-induced senescence and plays a role in promoting the senescence-associated secretory phenotype (SASP) by increasing NF-κB activity, which includes secretion of IL-6, IL-8, and granulocyte-macrophage colony-stimulating factor. Thus, our data (Figs. 4 and 5) shows that the synergistic effects of ALA on radiation-induced growth arrest may be sequentially due to (1) p38 activation, (2) NF-κB activation, (3) increased SASP expression, and finally, and (4) enhanced cellular senescence.
Mitochondria are known to cause cell damage by radiation-induced oxidative stress [25,26]. MitoSOX is a valid method for the detection of mitochondrial superoxide formation, which accurately represents the level of oxidative stress [27]. Oxidative stress and associated ROS can induce mitochondrial permeability transition and provide a signal leading to apoptosis [28]. In this study, MitoSOX and TMRM assays showed that RT combined with ALA increases mitochondrial oxidative stress and decreases mitochondrial membrane potential. This result indicates that oxidative stress, the main mediator of RT-induced cell damage, is further promoted by the synergistic effect of ALA.
ALA is a powerful antioxidant essential for aerobic metabolism. Although the body can synthesize ALA, the molecule can also be absorbed from nutritional supplements. It has been reported that ALA alone exhibits anti-cancer effects. It has been reported that ALA reduced cell viability/proliferation as well as lactate production, and increased apoptosis in neuroblastoma and breast cancer cell lines [5]. The molecule has also been proven to suppress STAT3-mediated MUC4 expression in a gastric cancer cell line [29]. Moreover, ALA had much stronger anti-cancer effects when it was combined with chemotherapeutic agents or RT. In colorectal cancer cells, ALA enhanced the cytotoxic effect of 5-fluorouracil [6]. Pretreatment with ALA prevented RT-induced epithelial-mesenchymal transition by inhibiting radiation-induced transforming growth factor β signaling and nuclear translocation of NF-κB in MCF-7 and MDA-MB-231 cells [8]. The evidence suggests that ALA not only is cytotoxic to cancer cells but also acts as a cancer radiation sensitizer while protecting normal tissues. However, the mechanism by which ALA sensitizes cancer cells to RT remains largely unexplored. The current study is the first report showing a new mode of action (senescence induction) of ALA beyond apoptotic cell death in RT-resistant breast cancer cells.
Our colleagues have demonstrated in previous studies that ALA acts as a potential radioprotector in various normal tissues during RT [9,10]. And, in this study, ALA showed synergistic effect as a radiosensitizer through cellular senescence in radioresistant breast cancer cells. From previous studies and this study, ALA appears to have an ideal dual effect, but additional experiments using other cancer cell lines or animal models are required, and well-designed clinical trials are also needed in the future for clinical use. We carefully suggest that ALA might be a potential novel pharmaceutical agent in cancer treatment and might contribute to a novel RT protocol for human cancer cells that are resistant to irradiation.