Introduction
In the 2016 World Health Organization (WHO) classification, histone 3 (H3) K27M-mutant diffuse midline glioma (DMG) was recognized as a distinct entity among high-grade gliomas [1]. DMG accounts for approximately 20% of pediatric glioblastomas, and its clinical characteristics have been described in detail in previous studies [2]. The midline locations of DMG tumors at presentation impede complete tumor removal, resulting in a poor survival outcome. Additionally, a previous study reported that H3 K27M-mutant DMG showed poorer survival than other grade IV gliomas that arose at the midline in a pediatric population [3]. However, growing understanding of the biology and descriptions of the outcomes of H3 K27M-mutant DMG is contributing to improvement of treatment outcomes.
DMGs are notable for harboring histone mutations, including H3F3A K27M, H3F3A G34R, and H3F3A G34V [4,5]. Genomic characterization of DMG in pediatric populations showed a high incidence of concurrent TP53 mutations and alterations in growth factor pathways [5]. In younger children in a mean age of 3 years with DMG, the ACVR1 somatic gain-of-function mutation was mainly found, resulting in the hyperactivation of bone morphogenetic protein signaling and ventralization of zebrafish embryos [6]. Additionally, genes involved in the phosphatidylinositol-3 kinase (PI3K) pathway were also altered. These genomic profiles facilitate the prognostications as well as precision treatment decisions of patients with DMG.
DMG is also found in adult patients with similar clinical features to those of pediatric patients [7,8]. However, because of its relatively rare incidence compared with pediatric populations, a comprehensive description encompassing clinical features and genomic profiles of adult DMG is still lacking. Furthermore, the clinical outcomes of palliative treatments for recurrent or refractory DMG have been scarcely described. Although precision medicine-guided treatment improved therapy recommendations using genomic and transcriptional profiling in advanced common cancers [9], precision medicine trials have been limited in rare cancers such as DMG. In this study, we described the clinical outcomes of adult patients with DMG and their genomic profiles through a retrospective analysis of 33 adult patients with DMG.
Materials and Methods
1. Patients and histology
We searched records for adult patients aged ≥ 18 years who were diagnosed with DMG involving the brain at Seoul National University Hospital between May 2005 and September 2019. DMG was defined as an infiltrative midline high-grade glioma with predominantly astrocytic differentiation and H3F3A or HIST1H3B/C mutation according to the 2016 WHO classification [1] by experienced pathologists (J.M.B. and S.-H.P). H3F3A K27M or G34V mutation was confirmed by the detection of K27M-mutant protein using an anti-histone H3.3 antibody (ABE419, Millipore, Temecula, CA) or targeted next-generation sequencing (NGS). Patients with an inconclusive diagnosis were excluded. The details on other histologic diagnosis methods including probes for immunohistochemistry (IHC) and fluorescent in situ hybridization are provided at Supplementary Methods.
Data including the patients’ demographics, radiographic findings, pathologic findings, diagnosis date, detailed treatments, initiation date of treatments, status of disease progression, and survival outcomes, were collected using the electronic medical records system.
2. Definition of clinical parameters and treatments
The surgical extent of DMG was classified into four groups based on the following: gross total resection (GTR) as complete resection of the mass and no visualized enhanced signal intensity in contrast-enhanced brain magnetic resonance imaging (MRI) obtained within 48 hours after surgery; near-total resection visualized as thin enhanced signal intensity in contrast-enhanced brain MRI obtained within 48 hours after surgery; subtotal resection as grossly remained tumor or visualized nodular enhancement in contrast-enhanced brain MRI obtained within 48 hours; biopsy only as biopsy without intent to tumor resection. We additionally evaluated T2 fluid attenuated inversion recovery (FLAIR) images to determine if there were any possible findings of residual tumors that were not identified by contrast-enhanced images.
The protocol used for concurrent chemoradiotherapy (CCRT) with temozolomide (TMZ) was either a standard 6-week [10] or hypofractionated 3-week interval based on age (≥ 70 years) or poor performance status [11]. The objective responses to radiotherapy with or without TMZ and palliative systemic treatments were evaluated according to the revised Response Assessment in Neuro-Oncology criteria [12]. Additionally, progression-free survival (PFS) was defined as the time to progression or death from treatment initiation and overall survival (OS) was defined as the time to death from diagnosis. We evaluated the association of the 6-month progression-free survival (PFS6) from radiotherapy initiation, a commonly used endpoint in glioblastoma, with the OS from diagnosis [13].
3. Targeted NGS profiles for adult DMG
Thirteen patients with available targeted NGS results were included. Genomic DNA was extracted from formalin-fixed paraffin-embedded tissue and was fragmented using a Covaris sonicator (Covaris, Woburn, MA). Agilent’s SureSelectXT Custom protocol was used to construct libraries (Agilent Technology, Santa Clara, CA), and MiSeqDx or NextSeq 550Dx (Illumina Inc., San Diego, CA) was used for sequencing.
The produced sequencing data were analyzed using the SNUH First Panel Analysis Pipeline. We performed quality control of the FASTQ file and further analyzed only the data that passed the criteria. Pair-end alignment to the hg19 reference genome was performed using BWA-men (v0.7.17) and GATK Best Practice [14,15].
After finishing the alignment step, an “analysis-ready BAM” was produced, and variants such as single-nucleotide variant (SNV), InDel, copy number variation (CNV), and translocation were detected using at least more than two analysis tools, including SNUH in-house and open-source software. GATK UnifiedGenotyper (v4.0.6.0), SNVer (v0.5.3), and LoFreq (v2.1.0) were used for SNV/InDel detection [15,16]; Delly (v0.7.8) and Manta (v1.4.0) for translocation discovery [17,18]; and THetA2 (v0.7) and CNVKit (v0.9.3) for purity estimation and CNV calling [19,20].
From those, we retained the variants meeting the following requirements: variant allele frequency ≥ 5%, reads supporting for alternative allele ≥ 10, reads supporting for alternative allele ≥ 10, and reads supporting each strand for alternative allele > 5 for SNV/InDel; copy number ≥ 6 and copy number ≤ 1 for CNV; split-reads supporting for alternative allele ≥ 10 for translocation. Other filtering criteria were applied using the default parameters of the analysis tools.
The detected variants were annotated by SnpEff (v4.3) using various databases such as RefSeq, COSMIC (v84), dbSNP (build 150), ClinVar (2018-06), and gnomAD (v2.0.1). The germline variant was filtered using the population frequency of these databases (> 1% population frequency) [21–26]. Finally, the variants were confirmed throughout a comprehensive review of a multidisciplinary molecular tumor board. The list of genes assessed by targeted NGS is provided in S1 Table.
4. Statistical analysis
Descriptive statistics were mainly used to demonstrate the results in this study. To describe and compare the survival outcome, Kaplan-Meier survival curves and log-rank survival analysis were used. Cox proportional hazard model for univariate analysis was used to determine clinical factors associated with OS in patients who received radiotherapy. Next, Cox proportional hazard model for multivariate analysis was used with age, gender and the factors with p-values less than 0.2. Logistic regression analysis was performed to evaluate whether a genomic alteration identified at least two samples by NGS affects PFS6 in patients who received CCRT. Ninety-five percent confidence intervals (CI) were calculated for survival analyses. Fisher exact test was used to compare categorical values. p-values less than 0.05 were considered as statistically significant. All statistical analyses were performed using R software ver. 3.5.1 (https://www.r-project.org).
Results
1. Patients and histologic findings
The demographics and histologic findings of patients with DMG are summarized in Table 1. Thirty-four adult patients were searched via an electronic medical record system, and 33 were confirmed to have H3-mutant DMG. One patient with an inconclusive diagnosis was excluded because of insufficient tumor tissue available for assessment. Thirty-two patients were confirmed by IHC, and one with H3F3A G34V mutation (S2 Fig.) was confirmed by targeted NGS. All the patients had lesions at the midline of the brain including the basal ganglia, thalamus, midbrain, pons, and medulla. All the patients had neurologic symptoms related to the lesion at the time of the first diagnosis. Dizziness and memory impairment were the most common symptoms (n=9, 27.3%, respectively).
IHC detected ATRX mutant protein in 10 of 31 patients (32. 2%). IDH1-mutant protein was observed in only one patient. IDH2 mutation was not observed. CDKN2A deletion and PTEN loss by fluorescence in situ hybridization were observed in six of 23 (26.1%) and eight of 23 (34.8%) patients, respectively. O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation was observed in one patient.
2. Genomic features of adult patients with DMG by targeted NGS
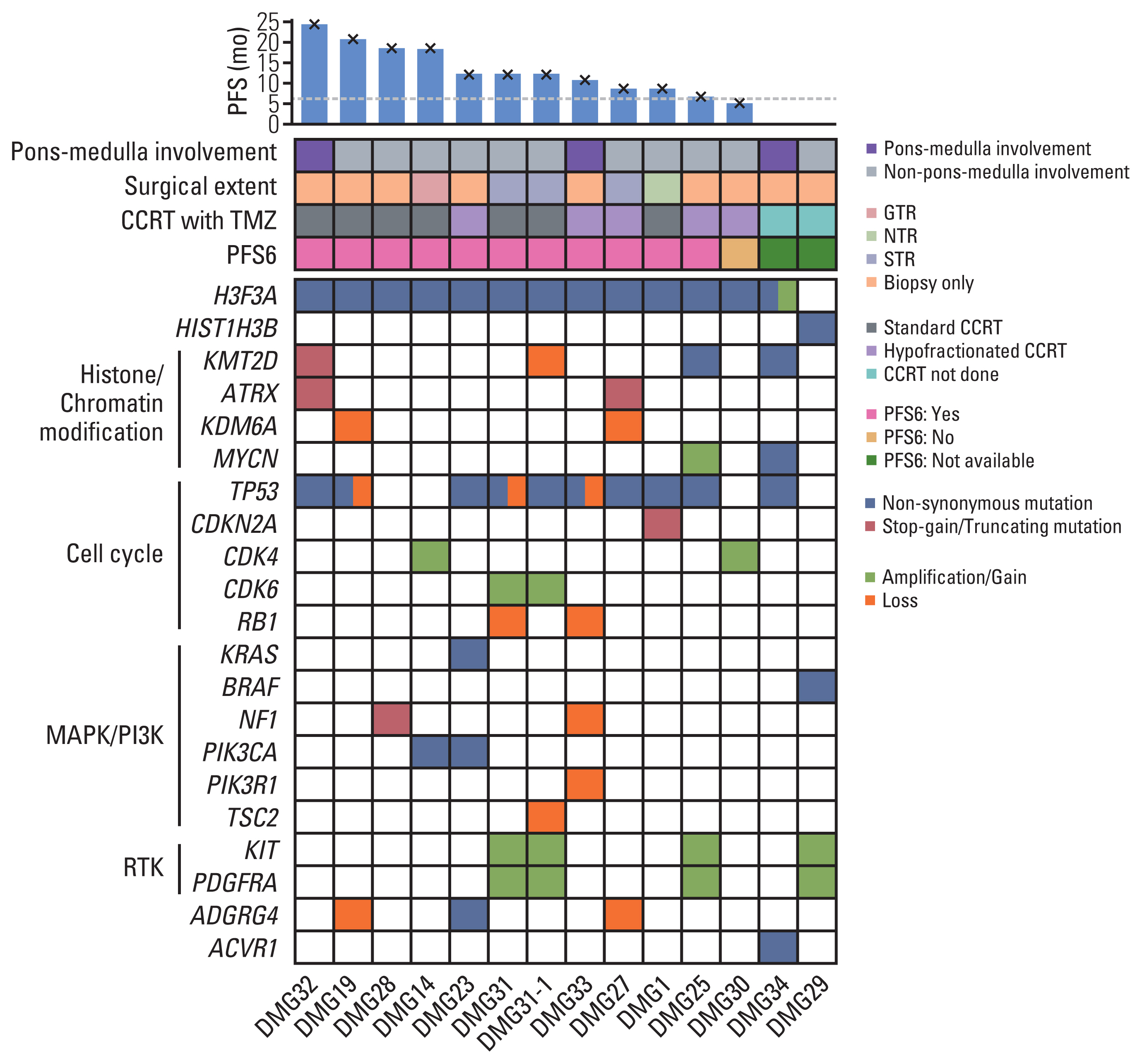
Thirteen patients had available targeted NGS data with a median tumor fraction of 85% (range, 20% to 90%) and a median average read depth of 1,016.3 (range, 649.7 to 2,066.5). All patients had H3 K27M mutation, among whom 12 had mutations at H3F3A and one had mutation at HIST1H3B. One patient had amplification of H3F3A. TP53 mutation was the most commonly altered gene (n=9, 69.2%). Eleven samples harbored alteration in cell cycle genes, including TP53, CDKN2A, CDK4, CDK6, and RB1. Alterations in mitogen-activated protein kinase (MAPK)/PI3K pathways, including KRAS, BRAF, NF1, PIK3CA, and PIK3R1, were observed in five patients (38.5%). Receptor tyrosine kinase amplification involving KIT and/or PDGFRA was observed in three patients (23.1%). ACVR1 and ATRX gene alterations were observed in one and two patients, respectively. IDH1 and IDH2 mutations were not observed in these 13 patients. None of the 13 patients had CDKN2A deletion or PTEN loss by targeted NGS. Structural variant was not identified in these patients. The genomic features of patients in this study are demonstrated in Fig. 1 (Detailed results are provided in S3 and S4 Tables).
One patient (DMG31) had paired targeted NGS data at the times of the first resection and re-surgery for relapsed tumor after adjuvant CCRT with TMZ (PFS of 13.3 months after the first resection). Although the latter sample harbored additional mutations in JUN and MAPK3, and loss of TSC2 (Fig. 1, S3 and S4 Tables), both samples showed similar genomic profiles with mutations in H3F3A and TP53 and amplifications in KIT, PDGFRA, and CDK6.
Comparisons with published mutational profiles are summarized in Table 2 [4,5,7,27]. TP53 mutations were frequently observed throughout the studies. ATRX and ACVR1 mutations were observed less commonly in our study than in other studies. Mutations in the MAPK/PI3K pathway were observed in few samples.
3. Survival outcomes of adult patients with DMG
The median OS from diagnosis was 21.8 months (95% CI, 13.2 to NA) (Fig. 2A), and 14 patients with involvement of the ponto-medullary area tended to have poor OS compared with 19 patients without involvement of the ponto-medullary area (median OS, 20.4 months [95% CI, 9.3 to not available (NA)] with involvement vs. 43.6 months [95% CI, 18.2 to NA] without involvement; p=0.07) (Fig. 2B). The 1-year and 3-year survival rates were 72.3% (95% CI, 57.6 to 90.7) and 33.4% (95% CI, 8.1 to 61.0), respectively.
Fourteen patients (42.4%) underwent surgical resection of tumors for initial treatment. While only three of 14 patients (21.4%) with ponto-medullary involvement underwent surgical resection, 11 of 19 patients (57.9%) without ponto-medullary involvement underwent surgical resection including three GTR cases. Surgical resection was not associated with prolonged OS in all patients (median OS, 21.9 months [95% CI, 13.2 to NA] for the resected group vs. 20.4 months [95% CI, 11.9 to NA] for the unresected group; p=0.84) (S5A Fig.). Additionally, GTR did not prolong the survival after surgery compared with non-GTR in patients who had undergone surgical resection (median PFS, 8.0 months [95% CI, 4.2 to NA] vs. 11.9 months [95% CI, 9.6 to NA]; p=0.40 [S5B Fig.] and median OS, 13.2 months [95% CI, 9.6 to NA] vs. 21.9 months [95% CI, 21.8 to NA]; p=0.60 [S5C Fig.]). Furthermore, one patient who achieved GTR and had no residual T2-high signal by T2 FLAIR images showed PFS after surgery of 19.4 months, which is the longest survival among the patients who achieved GTR.
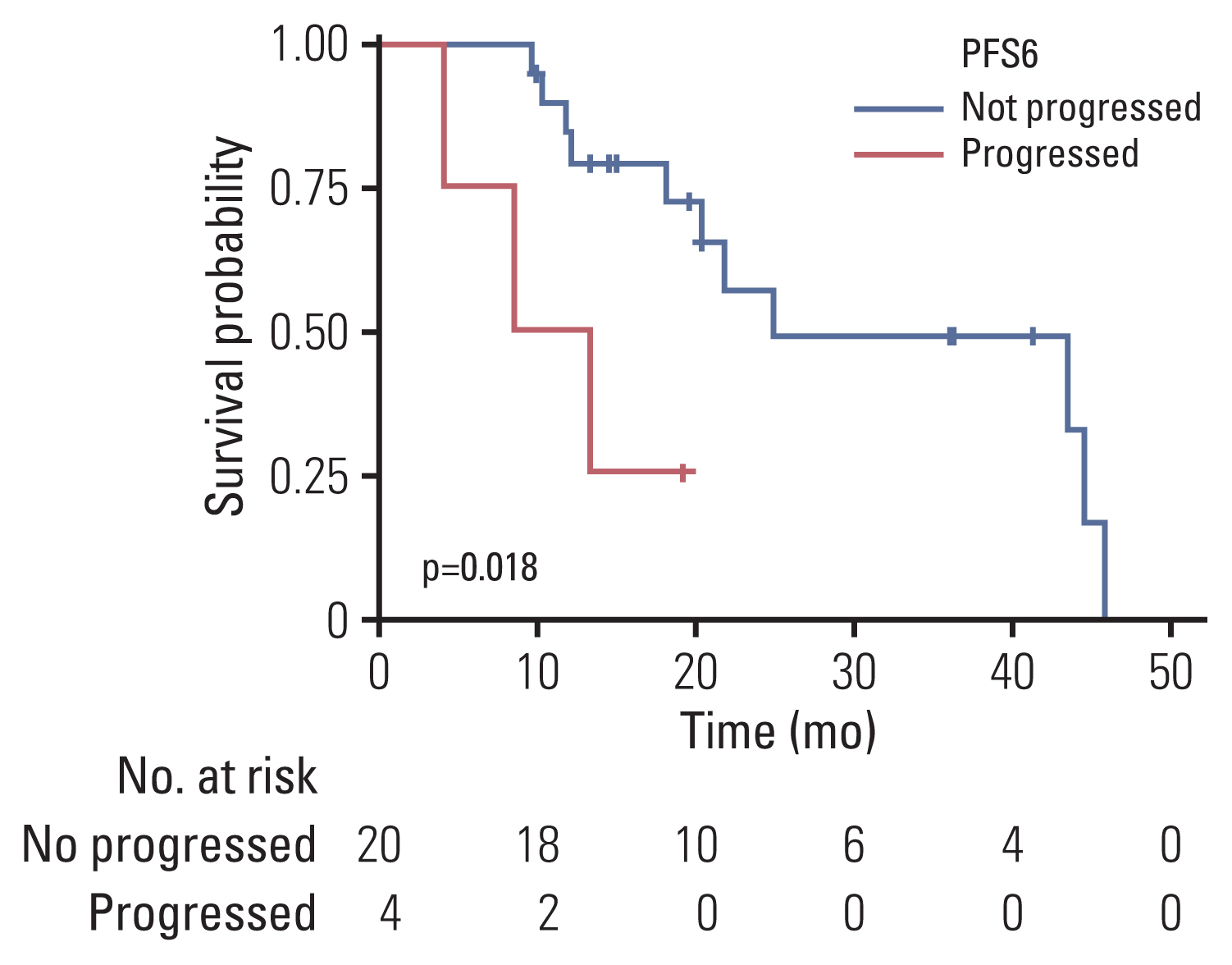
Twenty-four patients received radiotherapy: 20 and four patients received CCRT with TMZ and radiotherapy alone, respectively. Except for three patients who had received adjuvant chemoradiotherapy after GTR, the objective response rate of radiotherapy was 28.6% (n=6). The median PFS was 9.7 months (95% CI, 8.4 to 15.8), and the median OS was 24.9 months (95% CI, 18.2 to NA). The PFS6 rate was 83.3% (n=20; 95% CI, 69.7 to 99.7), and those without progression at 6 months showed significantly prolonged OS compared with the other four patients with progression at 6 months (median OS, 24.9 months [95% CI, 20.4 to NA] vs. 10.8 months [95% CI, 4.0 to NA]; p=0.02) (Fig. 3). The association of PFS6 and OS was consistent in multivariate analysis (hazard ratio, 0.12 [95% CI, 0.02 to 0.65]; p=0.01) (S6 Table). Comparisons of the treatment outcomes compared with other studies are summarized in Table 3 [8,28]. Tumor resection did not prolong PFS after radiotherapy (p=0.26) (S7A Fig.) and OS (p=0.61) (S7B Fig.). Additionally, GTR was not associated with prolonged PFS after radiotherapy in patients who had undergone resection (p=0.64) (S7C Fig.).
ATRX mutation was not associated with PFS compared with ATRX wild type (median PFS, 11.4 months [95% CI, 6.8 to NA] vs. 10.7 months [95% CI, 8.4 to 18.4]; p=0.86). Neither CDKN2A deletion nor PTEN loss was associated with PFS (p=0.41 and p=0.22, respectively). Additionally, ATRX mutation, CDKN2A deletion; and PTEN loss were not associated with OS in the patients who had received CCRT with TMZ (p=0.74, p=0.93, and p=0.84, respectively). In addition, genomic alterations by NGS were not associated with PFS6 (S8 Table).
4. Efficacy and survival outcomes of salvage systemic therapies
Eleven patients had received salvage systemic therapies. The median OS from the initiation of systemic therapy was 8.3 months (95% CI, 6.1 to NA) in these patients. Six of these patients had surgical resection, and GTR was achieved in two patients. Bevacizumab was administered in seven patients as first-line salvage therapy after CCRT with TMZ, three of whom received irinotecan as combination.
The objective response rate, median PFS, and median OS after bevacizumab-based regimen were 28.6%, 1.6 months (95% CI, 1.4 to NA) (S9A Fig.), and 6.1 months (95% CI, 6.1 to NA), respectively. The two responders showed longer OS rates (19.9 and 11.2 months) (S9B Fig.).
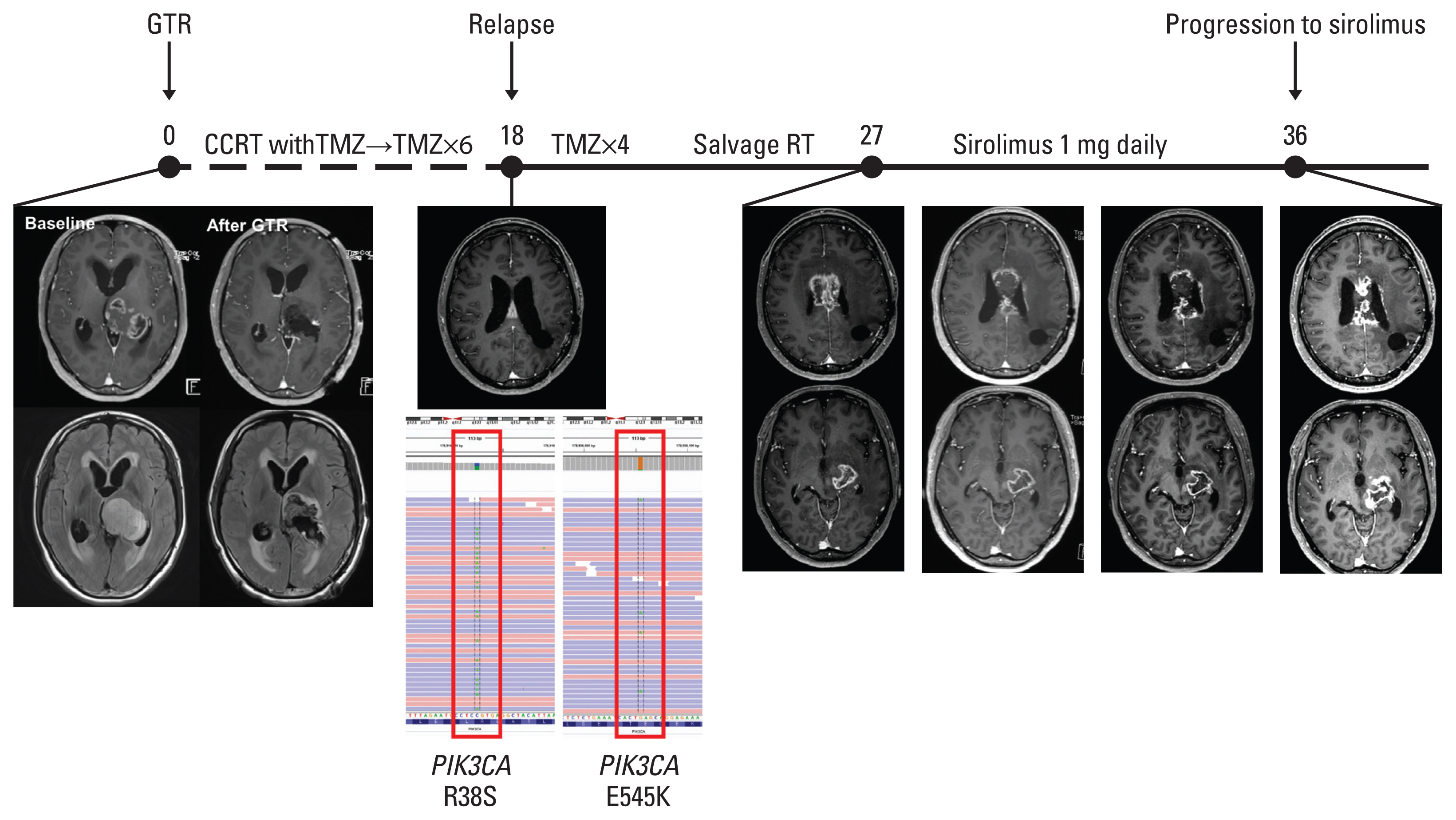
Two patients received matched therapies in clinical trials: patient DMG14 with concurrent PIK3CA R38S (variant allele frequency [VAF] of 57.8%) and E545K (VAF of 7.2%) mutations had received sirolimus (NCT02688881); and patient DMG23 with KRAS G12A mutation (VAF 24.0%) had received pan-RAF plus MEK inhibitors. Patient DMG14 who experienced rapid progression before the administration of sirolimus showed stable disease to sirolimus with a PFS of 8.4 months (Fig. 4). Patient DMG23 showed a metabolic response to pan-RAF plus MEK inhibitors at 1 month along with metabolic improvement at the ipsilateral hemisphere, although she stopped investigational products because of a poor general condition, resulting in an OS of 5.6 months (S10 Fig.).
Discussion
Despite growing attention concerning pediatric DMG in various studies, the data on adult patients with DMG are only described in few studies [7,8]. Here, we described the demographics of adult DMG patients and clinical outcomes of definitive treatments and palliative systemic treatments. Our study demonstrated that patients may receive matched therapies based on genomic profiles, implying potential feasibility of individualized approach for rare population of adult DMG.
As a driver mutation for glioblastoma, H3F3A mutation was first discovered by whole-exome sequencing of pediatric glioblastoma samples [5]. H3F3A K27M mutation enhances neural stem cell self-renewal and cooperates with the activating platelet-derived growth factor receptor α mutant and Trp53 loss to accelerate the development of diffuse brainstem gliomas [29]. We showed that a high incidence of TP53 mutation, which is consistent with several previous studies (Table 2) [4,5,7,27]. H3F3A gain-of-function mutation also inhibits PRC2 activity to alter epigenetic silencing and increase H3K27 acetylation [30]. Through these mechanisms, DMGs reprogram the methylation landscape and gene expression to harbor distinct genomic, methylation, and transcriptomic profiles from other glioblastomas [4,5,31]. The distinct features of DMG from other glioblastomas also include an extremely rare frequency of IDH1/2 mutation [8,32], a finding that was also observed in our study. Although H3F3A G34V-mutant glioma is primarily located at the cerebral hemisphere [4], our DMG34 case with H3F3A G34V mutation showed a midline location and TP53 mutation (S2 Fig.), similar to typical H3F3A K27M-mutant DMG cases.
We failed to demonstrate the prognostic significance of genomic profiles such as ATRX mutation, CDKN2A deletion, and PTEN loss. This finding might be attributable to the small number of patients in our study as well as the poor prognosis of DMG [3,4].
The clinical outcomes of our adult patients with DMG were compared with those of two previous studies of adult patients with DMG (Table 3) [8,28]. Although the resection rate including GTR (9.1%) was higher in our study, no survival differences were found between the studies [3]. These results could have been attributed to the midline location of DMGs and their infiltrative nature being difficult for surgeons to achieve complete resection. By contrast, the survival outcomes were associated with the sensitivity to radiotherapy defined as PFS6 after radiotherapy in our study. Patients without progression at 6 months after radiotherapy survived longer than those with progression at 6 months after radiotherapy, suggesting the important role of radiotherapy in adult patients with DMG.
Although patients who received palliative systemic treatments had a median OS of 8.3 months, a few responders seemed to have a prolonged OS. Additionally, two of 13 patients who had targeted NGS received matched therapies, including pan-RAF plus MEK inhibitors to target KRAS mutation and sirolimus to target PIK3CA mutation, suggesting the potential approach for precision medicine-based therapy. Regarding pathogenic PIK3CA mutations, previous study has demonstrated the potential efficacy of sirolimus in refractory solid cancers [33]. A recent high-throughput combination drug screening strategy identified the potential therapeutic combination of the multi-histone deacetylase inhibitor panobinostat and proteasome inhibitor marizomib against the preclinical DMG model [34]. In addition, inhibition of EZH2, an enzymatic subunit of polycomb repressive complex 2, reduced tumor growth in the H3 K27M-mutant mouse model and patient-derived cell lines, suggesting an epigenetic modifier as a potential therapeutic target for H3 K27M-mutant patients with DMG [35,36]. However, these preclinical therapeutic strategies should be validated in patients with treatment-refractory or relapsed DMG.
The limitations of our retrospective study include the small number of patients with adult DMG as well as limited number of targeted NGS. Therefore, genomic demographics and their clinical associations are not fully elucidated in this study. In addition, there seemed modest benefits of matched investigational drugs in two patients.
In conclusion, our study demonstrated that a progression-free survival at 6 months after radiotherapy significantly improved survival in adult patients with DMG while surgical extent did not improve survival, suggesting the important role of radiotherapy in patients with adult brain DMG. Additionally, targeted sequencing revealed a high frequency of concurrent TP53 mutation in combination with H3F3A or HIST1H3B mutation. Considering two patients with MAPK-PI3K alterations received matched therapy, a precision medicine-guided treatment approach might be feasible in adult patients with brain DMG. Future efforts should be directed toward targeting histone/chromatin modification or co-targeting histone/chromatin modification and other pathways based on the DMG genome in a basket or platform trial.