Introduction
Pim kinases are a class of constitutively active serine/threonine kinases consisting of three highly homologous members: Pim-1, Pim-2, and Pim-3. These factors were discovered as proviral insertion sites of the Moloney murine leukemia virus associated with the development of T-cell lymphomas. Members of this kinase family have been implicated in several biological processes, including cell survival, proliferation, and differentiation [1]. Pim kinase expression is mainly regulated by transcription factors, such as Janus kinase/Signal transducer and activator of transcription (JAK/STAT); the nuclear factor κB, pathway; and ubiquitylation with subsequent proteasomal degradation at the post-translational level [2].
Pim kinases modulate cell proliferation by phosphorylating cell cycle regulators, such as p21, p27, Cdc25A, and Cdc-25C, and mediate survival signal pathways by phosphorylating the Bcl-2 antagonist of cell death (Bad). Furthermore, these kinases have been found to induce phosphorylation of 4E-binding protein 1 (4E-BP1), which permits protein synthesis by 5′ cap-dependent translation [3]. Mechanistic studies have revealed that high expression levels of Pim are associated with tumorigenesis and cancer progression.
The expression level of each Pim isoform varies among tumor types [3]. More recently, the Pim-1 and Pim-3 levels have been found to be increased and predictive of poor prognosis in gastric cancer [4,5]. In particular, there is a relationship between aberrant expression of Pims and aggressive types of gastric cancer. Previous studies have reported that Pim-3 expression is greater in a tissue section of gastric adenoma compared to non-cancerous mucosa and is higher in intestinal-type gastric carcinoma than in diffuse-type gastric carcinoma. Interestingly, Pim-3 is expressed at a higher rate in metastatic sites than in primary sites of gastric carcinoma, and consequently, there is a lower survival rate of patients with Pim-3-positive gastric cancer [5]. Furthermore, several studies have also reported that the JAK/STAT signaling pathway, which directly modulates the expression levels of Pim kinases, is hyper-activated in gastric cancer [6]. Thus, Pim kinases have become a promising therapeutic target for the treatment of gastric cancer. However, it is well-known that Pim isoforms have compensatory effects, allowing them to maintain each other’s functions [1]. For this reason, most Pim inhibitors are designed to inhibit all Pim isoforms; however, the effects of pan-Pim inhibitors and their underlying mechanisms have not been adequately explained in gastric cancer.
Dysregulation of the phosphatidylinositide 3-kinase (PI3K)/Akt pathway is commonly found in many types of human cancer, including gastric cancer [7]. Therefore, inhibition of this pathway is an attractive therapeutic target for treating gastric cancer. Although small molecule inhibitors that target Akt have been developed, due to the activation of compensatory signaling pathways following Akt inhibition, there have been many efforts to improve the effects of Akt inhibitors through combination strategies [8-10]. Previous studies have reported that resistance to Akt inhibition is mainly induced by mammalian target of rapamycin (mTOR) activation [11]. There have been many cases suggesting a treatment strategy of using mTOR and Akt inhibition; however, tolerability issues have been limiting. Pim and Akt kinases possess independent signaling pathways, but overlap in several areas. For example, PRAS40 activity is regulated by Pim kinases independent of Akt activation, but both directly regulate proapoptotic molecules, such as the Bcl-2 protein Bad [12,13]. Therefore, an inhibitory combination of Pim and Akt is expected to overcome Akt resistance by regulating mTOR activity. Furthermore, up-regulation of receptor tyrosine kinases by Pim induce resistance to Akt inhibitors [14]. Therefore, simultaneous inhibition of Pim and the Akt pathway could be a potent anticancer therapeutic strategy [15].
In the present study, the antitumor activity of the pan-Pim inhibitor AZD1208 [16], as well as its underlying mechanism, was evaluated in a large panel of gastric cancer cell lines. Subsequently, we explored the role of AZD1208 as a sensitizer when administered in combination with the Akt inhibitor AZD5363. Here, we show for the first time that treatment with AZD1208 alone exerts cytotoxic effects by inducing autophagy. This is the first report to show that inhibition of Pim kinases can suppress the growth of gastric cancer cells and that the DNA damage response is related to the sensitivity to AZD1208. We also show the therapeutic significance of dual inhibition of the Pim and Akt pathways, which synergistically decrease cell viability and improve drug efficacy in human gastric cancer cells.
Materials and Methods
1. Reagents
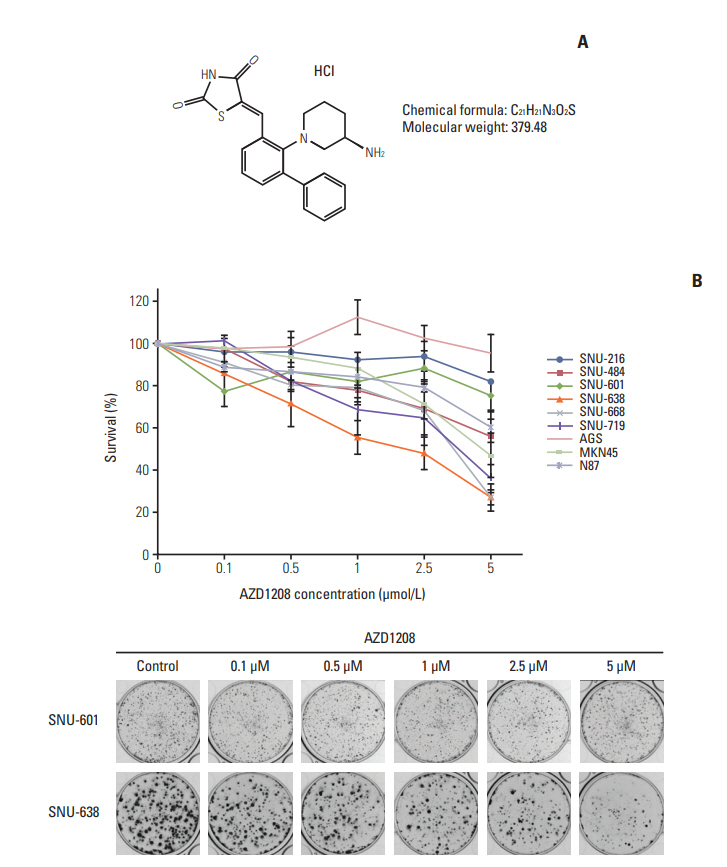
AZD1208, a Pim inhibitor [16,17], and AZD5363, an Akt inhibitor [18], were kindly provided by AstraZeneca (Macclesfield, Cheshire, UK). The chemical structure of AZD1208 is shown in Fig. 1A. 3-Methyladenine (3-MA) was purchased from Sigma-Aldrich (St. Louis, MO), and N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone (z-VADfmk) was obtained from R&D Systems (Minneapolis, MN).
2. Cell lines and cell culture
Human gastric cancer cell lines (SNU-1, -5, -16, 216, -484, -601, -620, -638, -668, -719, AGS, MKN45, KATO-III, and N87) were purchased from the Korean Cell Line Bank (Seoul, Korea). The identities of the cell lines were authenticated by DNA fingerprinting analysis [19]. All cell lines were banked and passaged for less than 6 months before use. The cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific Inc., Waltham, MA) supplemented with 10% fetal bovine serum (Welgene, Daegu, Korea) and 10 μg/mL gentamicin (Cellgro, Manassas, VA) at 37°C and 5% CO2.
3. Cell growth inhibition assay
Cells (2-3×103 in 100 μL/well) were seeded in 96-well plates (TPP, Trasadingen, Switzerland) and incubated overnight at 37°C and 5% CO2. The cells were then exposed to increasing concentrations of AZD1208 (range, 0.001 to 10 μmol/L) or dimethyl sulfoxide (DMSO; Sigma-Aldrich) for 5 days. After treatment, 50 μL of a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolim bromide (MTT) solution (Sigma-Aldrich) was added to each well, and the plates were incubated for 4 hours at 37°C before the media was removed. After dissolving formazan crystals with 150 μL of DMSO, the absorbance of each well was measured at 540 nm with a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). The absorbance and IC50 of the Pim inhibitor were analyzed using SigmaPlot software (SPSS Inc., Chicago, IL). Six replicate wells were included for each analysis, and at least three independent experiments were performed.
4. Colony formation assay
For the colony formation assay (CFA), cells were seeded into 6-well plates and incubated for 48 hours at 37°C in 5% CO2. Next, the cells were treated with various concentrations (0.1, 0.5, 1, 2.5, and 5 μmol/L) of AZD1208 alone or in combination with 100 nmol/L AZD5363 every 3 days. The cells were cultured until colonies formed (14 days). The colonies were stained with a 0.1% Coomassie blue solution (Sigma-Aldrich) and counted using a GelCount automatic plate scanner (Oxford Optronics GelCount, Oxford, UK). The cell survival rate and IC50 of AZD1208 were determined using SigmaPlot software (SPSS Inc.).
5. Pyrosequencing analysis
Target-specific primers were designed for pyrosequencing analysis as follows: PIK3CA-E453K forward, 5′-biotin-GCTTTGAATCTTTGGCCAGTAC-3′; PIK3CA-E453K reverse, 5′-TCCAGTAACACCAATAGGGTTCAG-3′; PIK3CA-E542K and -E545K forward, 5′-biotin-CAGCTCAAAGCAATTTCTACACG-3′; PIK3CA-E542K and -E545K reverse, 5′-GCACTTACCTGTGACTCCATAGAA-3′; PIK3CA-H1047R forward, 5′-biotin-TTCGAAAGACCCTAGCCTTAGAT-3′; PIK3CAH1047R reverse, 5′-TGCTGTTTAATTGTGTGGAAGATC-3′. For PIK3CA-E453K, E542K, E545K, and H1047R, the pyrosequencing primers 5′-AGGGTTCAGCAAATCT-3′, 5′-TTCTCCTGCTCAGTGAT-3′, 5′-CCATAGAAAATCTTTCTCCT-3′, and 5′-TGCTGTTTAATTGTGTGGAAGATC-3′were used, respectively. Single-stranded biotinylated PCR products were processed for pyrosequencing analysis according to the manufacturer’s standard protocol (PyroMark Q96 ID, Qiagen, Hilden, Germany).
6. Reverse transcription–polymerase chain reaction and real-time polymerase chain reaction
Total RNA was isolated using TRI reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s instructions. cDNA was synthesized by reverse transcription polymerase chain reaction (RT-PCR) with ImProm-II reverse transcriptase (Promega, Madison, WI) and amplified using AmpliTaq Gold DNA polymerase (Applied Biosystems, Carlsbad, CA) with gene-specific primers. Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted using an iCycler iQ detection system (Bio-Rad Laboratories, Inc., Hercules, CA) with SYBR Green. Data for all samples were normalized relative to actin cDNA. The sequences of the primers used for RT-PCR and qRT-PCR are listed in S1 Table. cDNA was synthesized at least three times from three independent sets of samples, and all PCR analyses were performed in triplicate.
7. Western blot analysis
Proteins were extracted, and equal amounts of protein were separated on 5%-15% sodium dodecyl sulfate–polyacrylamide gels as previously described [20]. Separated proteins were transferred onto nitrocellulose membranes, and the blots were probed with primary antibodies (1:1,000 dilution) overnight at 4°C. Antibodies specific for the following factors were used: Pim-1 (sc-13513, Santa Cruz Biology Technology, Santa Cruz, CA), Pim-2 (CST 4730, Cell Signaling Technology, Beverly, MA), Pim-3 (CST 4165), phosphorylated (p)Bad Ser112 (CST 9296), Bad (CST 9292), p4EBP1 Ser65 (CST 9451), 4EBP1 (CST 9452), pS6K (CST 2211), S6K (CST 2217), Bcl-xL (sc-1690), Mcl-1 (sc-819), PARP (BD 556-494), caspase-3 (CST 9662), light chain 3B (LC3B; CST 3868), Beclin-1 (ab51031), pATM Ser1981 (CST 4526), ATM (sc-239-21), pChk2 Thr68 (CST 2661), Chk2 (sc-5278), pAkt Ser473 (CST 9271), Akt (CST 9272), pPRAS40 Thr246 (CST 2997), PRAS40 (CST 2691), and α-tubulin (Sigma-Aldrich). Antibody binding was detected using an enhanced chemiluminescence system according to the manufacturer’s protocol (Amersham Biosciences, Piscataway, NJ).
8. Cell-cycle analysis
Cells treated with AZD1208 were harvested, fixed with cold 70% ethanol, and then stored at –20°C for at least 24 hours. Next, the cells were washed in phosphate buffered saline (PBS), incubated with 10 μg/mL RNase A (Sigma-Aldrich) at 37°C for 20 minutes, and stained with 20 μg/mL propidium iodide (Sigma-Aldrich). The DNA levels of the cells (10,000 cells per group) were quantified using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA).
9. Annexin V binding assay
After the cells were exposed to AZD1208, the degree of apoptosis was assessed using the Annexin V binding assay according to the protocols of the manufacturer (BD PharMingen, San Jose, CA). The harvested cell suspension was then incubated with Annexin V for 15 minutes at room temperature in the dark and analyzed by flow cytometry.
10. Immunofluorescence assay
Cells were plated on 0.01% poly-L-Lysine (Sigma Aldrich)–coated coverslips, transfected with GFP-labeled LC3B and treated with 1 or 5 μM AZD1208. After 5 days, the coverslips were rinsed once in PBS (37°C), fixed in 3.7% paraformaldehyde for 10 minutes, permeabilized with PBS-T (0.5% TritonX-100 in PBS) for 5 minutes, and incubated with primary antibodies for 24 hours at 4°C. The primary antibodies used in this study included anti-LC3B, -pChk2, and anti-γH2AX at a dilution of 1:50. The coverslips were rinsed three times for 10 minutes in PBS, followed by incubation with the appropriate fluorophore-conjugated secondary antibody (Invitrogen, Carlsbad, CA). The cells were counterstained with DAPI (300 nM, Invitrogen), and the coverslips were mounted onto slides using Faramount aqueous mounting medium (Dako, Glostrup, Denmark). Immunofluorescence was visualized using a Nikon A1 confocal laser scanning microscope (Nikon, Tokyo, Japan).
11. In vivo study
The animal experiments were carried out at the animal facility of Seoul National University (Seoul, Korea) according to the institutional guidelines with prior approval from the Institutional Animal Care and Use Committee. Six-week-old female BALB/c nude mice purchasing from Central Lab Animal Inc. (Seoul, Korea) were used to test the in vivo activities of AZD1208. The mice were injected subcutaneously in the right flank with 7×107 of SNU-638 cells in 100 μL of PBS. After implantation of the tumor cells, the tumor sizes were measured every other day using calipers, and the body weight of each mouse was determined twice per week. The mice were randomly divided into two groups (five mice per group) when tumor volumes reached 200 mm3, and 45 mg/kg of AZD1208 were administered via oral gavage once daily for 28 consecutive days. The control group was treated with vehicle alone (1 mM histidine, 130 mM Glycine, 5% sucrose in water). Tumor volume was examined not only during treatment but also after treatment was ceased and calculated using the following formula: (width2×height)/2. The mice were sacrificed with CO2 at the end of the observation period, and tumors were excised for further analysis.
12. Immunohistochemistry
Paraffin-embedded xenograft tumor tissues were deparaffinized with xylene and rehydrate with graded ethanol. Immunohistochemistry studies for Ki-67 were conducted by using the anti-rabbit polyclonal antibody against Ki-67 (dilution of 1:100, GeneTex, Irvine, CA) and a terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling assay performed to measure the apoptosis of xenografts using ApopTag In situ Apoptosis Detection Kit (Chemicon International, Temecula, CA) following the manufacturer’s protocol.
Results
1. AZD1208 suppresses tumor growth in gastric cancer
To determine the effects of AZD1208 treatment on human gastric cancer cell growth, we first treated each cell line with various concentrations of AZD1208 for 120 hours. Cell survival was then measured via the MTT assay (S2A Fig.). AZD1208 had a minimal anti-proliferative effect on most of the cell lines up to a concentration of 1 μM. At 10 μM, AZD-1208 suppressed the proliferation of N87 and MKN45 cells by approximately 40%; however, no obvious growth inhibition was observed during the early time points of the cell viability assay. Based on our data and previous reports stating that Pim kinases promote cell cycle progression and evasion of apoptosis signals, we had predicted a lower proliferation rate and active cell death signals when Pim kinases were disrupted [21,22]. We confirmed this prediction in longterm colony formation assays. We observed that AZD1208 treatment affected cell proliferation in gastric cancer cell lines (Fig. 1B, S2B Fig.), and several additional cell lines (i.e., SNU-484, -638, and -719) showed greater dose-dependent sensitivity to AZD1208 treatment than other cell lines (Table 1); SNU-638 cells were the most sensitive to AZD1208 compared to other cell lines, and SNU-601 cells were the most resistant. Based on the results obtained, SNU-638 and SNU-601 cells were selected for further study. In addition, AZD1208 significantly delayed tumor growth in a SNU-638 xenograft model. The doubling time of tumor volume with vehicle treatment was 17 days, while the time to 2-fold increase of tumor volume with AZD1208 treatment was observed after 31 days, which supports the delay of tumor growth by AZD1208 treatment. Furthermore, AZD1208 treated mice showed lower Ki-67 expression, suggesting lower proliferation ability compared with non-treated mice. However, there was no meaningful increase in apoptosis in AZD1208 treated mice (S3 Fig.). These data demonstrated the antitumor effects of AZD1208 in a gastric cancer model.
2. Expression of Pim isoforms is not correlated with the response to AZD1208
To determine whether cell line sensitivity was correlated with Pim isoform expression, we analyzed the mRNA and protein expression levels of Pim-1, Pim-2, and Pim-3 in a panel of 10 gastric cancer cell lines. To assess mRNA expression, we performed RT-PCR and qRT-PCR analyses (S4 Fig.). Differential expression of Pim family members was observed, with Pim-3 expression mostly up-regulated as previously reported [5]. Additionally, we observed heterogeneous protein expression of the Pim isoforms in each cell line (Fig. 2). These data suggest that the basal levels of Pim-1, Pim-2, and Pim-3 mRNA and protein did not correlate with responses to AZD1208.
Interestingly however, we found that SNU-601 and AGS cells harboring PIK3CA mutations showed no sensitivity to AZD1208 (Table 2), raising the possibility that PI3K/Akt pathway activation may promote resistance to AZD1208.
3. AZD1208 inhibits phosphorylation of Pim kinase substrates
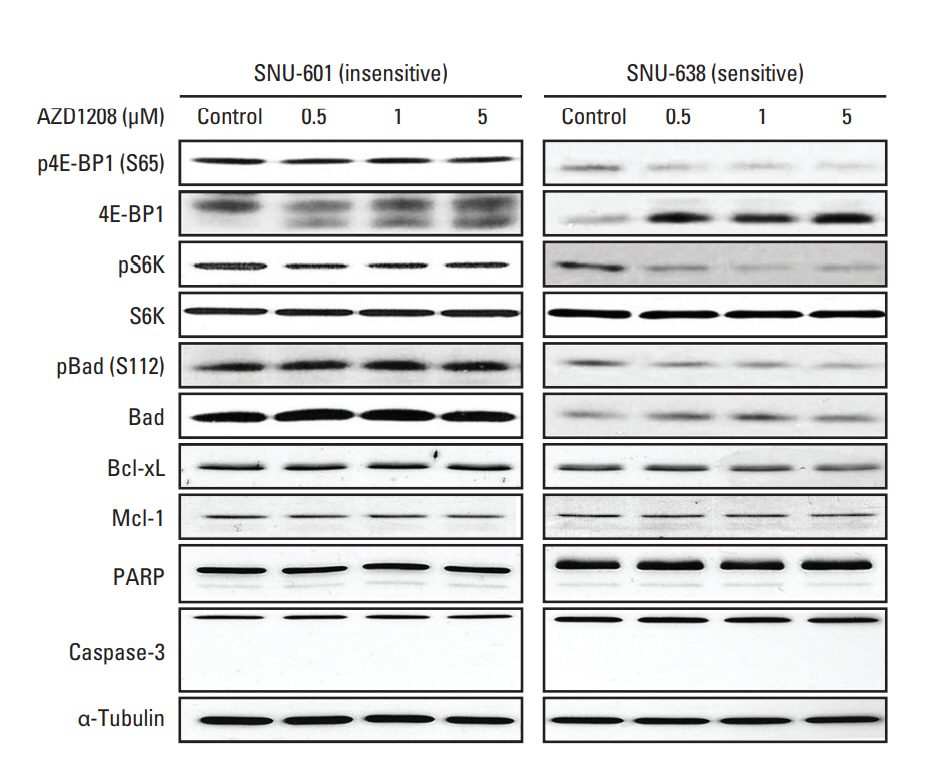
To determine whether AZD1208 was capable of inhibiting phosphorylation of proteins downstream of Pim kinases, we measured the phosphorylation and total protein expression levels of these downstream molecules by western blot analysis [1,3]. A significant dose-dependent reduction of phosphorylation of the Pim substrates Bad and 4E-BP1 in SNU-638 cells was observed. In addition, phosphorylation of S6K, another downstream effector of mammalian target of rapamycin complex 1 (mTORC1), was also markedly down-regulated. On the other hand, there were no changes in the levels of pro-survival proteins, such as Bcl-xl and Mcl-1. Moreover, we did not see any increase in the protein levels of PARP cleavage and caspase-3 activity via immunoblotting or in apoptotic cells via annexin V assays after AZD1208 treatment (Fig. 3, S5 Fig.).
Major proteins of oncogenic proliferation pathways, such as Akt or Erk, and modulators of Pim expression, such as the Jak/STAT pathway, were not affected by AZD1208 treatment in gastric cancer cells (data not shown). However, the influence of AZD1208 on downstream substrates of Pim kinase and downstream signals of mTORC1 correlated with drug sensitivity. Collectively, these data suggest that downstream molecules of Pim kinase can be regulated by AZD-1208, but apoptosis induction or inhibition of proliferative signaling is not responsible for the anti-tumor effects of AZD1208.
4. AZD1208 induces autophagic death of gastric cancer cells
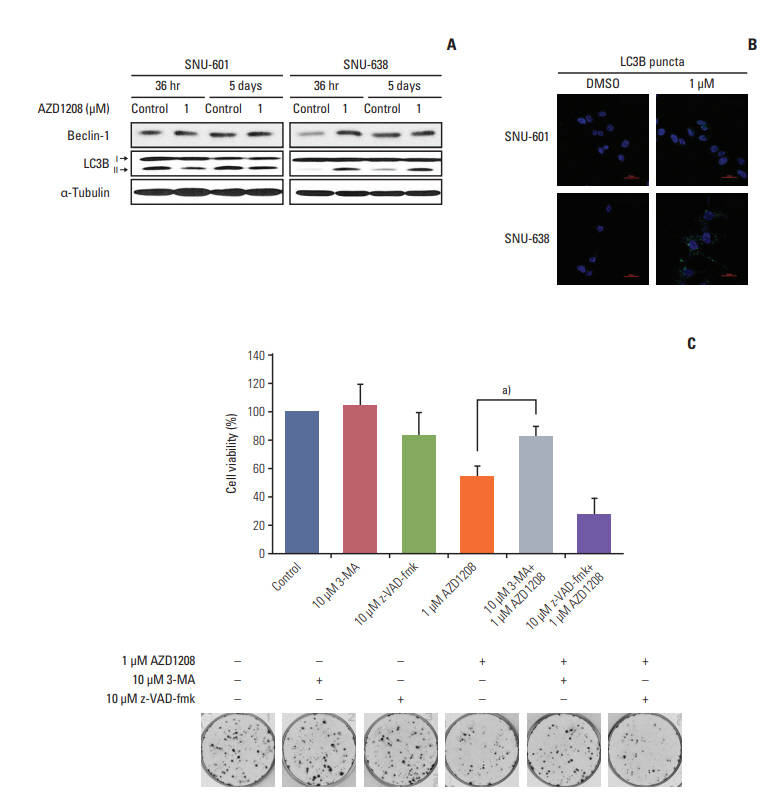
Since we found that AZD1208 did not induce apoptosis, we determined whether autophagy (or type II programmed cell death) was related to the cytotoxic effects of AZD1208. First, we measured the expression levels of the well-known autophagy markers LC3B and Beclin-1 following AZD1208 treatment (Fig. 4A). LC3B is cleaved to form LC3B-II during autophagy; thus, conversion of LC3B-I to LC3B-II is associated with autophagosome formation. Interestingly, AZD1208 treatment induced up-regulation of LC3B-II expression at both time points in SNU-638 cells, but not in SNU-601 cells. By contrast, Beclin-1 expression was slightly up-regulated at 36 hours in SNU-638 cells, but no visible changes were observed after 5 days of exposure.
Next, we performed an immunofluorescence study to further confirm the role of AZD1208 on the induction of autophagy. Both SNU-601 and SNU-638 cells were transfected with a plasmid encoding GFP-tagged LC3B. Fluorescence microscopy was then performed to monitor GFP-tagged LC3 expression in both sensitive and resistant cells (Fig. 4B). SNU-638 cells clearly showed re-localization of GFP-LC3B from a diffuse pattern into a punctate pattern corresponding to autophagosome formation following AZD1208 treatment. By contrast, GFP-LC3B localization was cytosolic and diffuse in SNU-601 cells, regardless of drug treatment.
To determine whether AZD1208 sensitivity was a direct result of autophagy and not apoptosis, we performed CFAs to measure the growth of SNU-638 cells in the presence of the autophagy inhibitor 3-MA and caspase inhibitor z-VADfmk (Fig. 4C). 3-MA treatment in SNU-638 cells effectively rescued the AZD1208-induced decrease in cell viability. However, z-VAD-fmk treatment did not result in restoration of cell viability but instead slightly decreased cell viability. These data demonstrate that autophagic cell death induced by AZD1208 has antitumor effects on gastric cancer cells.
5. Regulation of the DNA damage response is associated with AZD1208 sensitivity
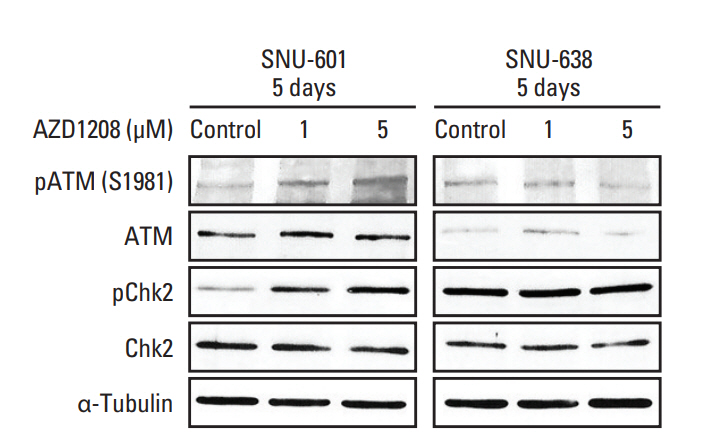
A role of Pim kinases in repairing DNA damage has been reported [23]. We therefore determined whether AZD1208 can affect the DNA damage repair (DDR) pathway via western blot analysis. Intriguingly, ATM phosphorylation was up-regulated in a dose-dependent manner in insensitive SNU-601 cells along with Chk2 phosphorylation (Fig. 5). Consistent with these findings, we also observed that Chk2 expression was highly activated in the nuclei of SNU-601 cells, but not those of in SNU-638 cells (S6 Fig.). Data from these experiments revealed that AZD1208 treatment induced DNA damage and hyper-activation of the DDR in SNU-601 cells correlated with increased resistance to AZD1208. Depletion of Pim kinases can lead to DNA damage accumulation [21,23], and regulation of the DDR may be related to drug sensitivity [24]. These results suggest that increased activity of the DDR system could be a mechanism underlying AZD1208 resistance.
6. Combined treatment of AZD1208 with an Akt inhibitor enhances antitumor effects and overcomes drug resistance in gastric cancer cells
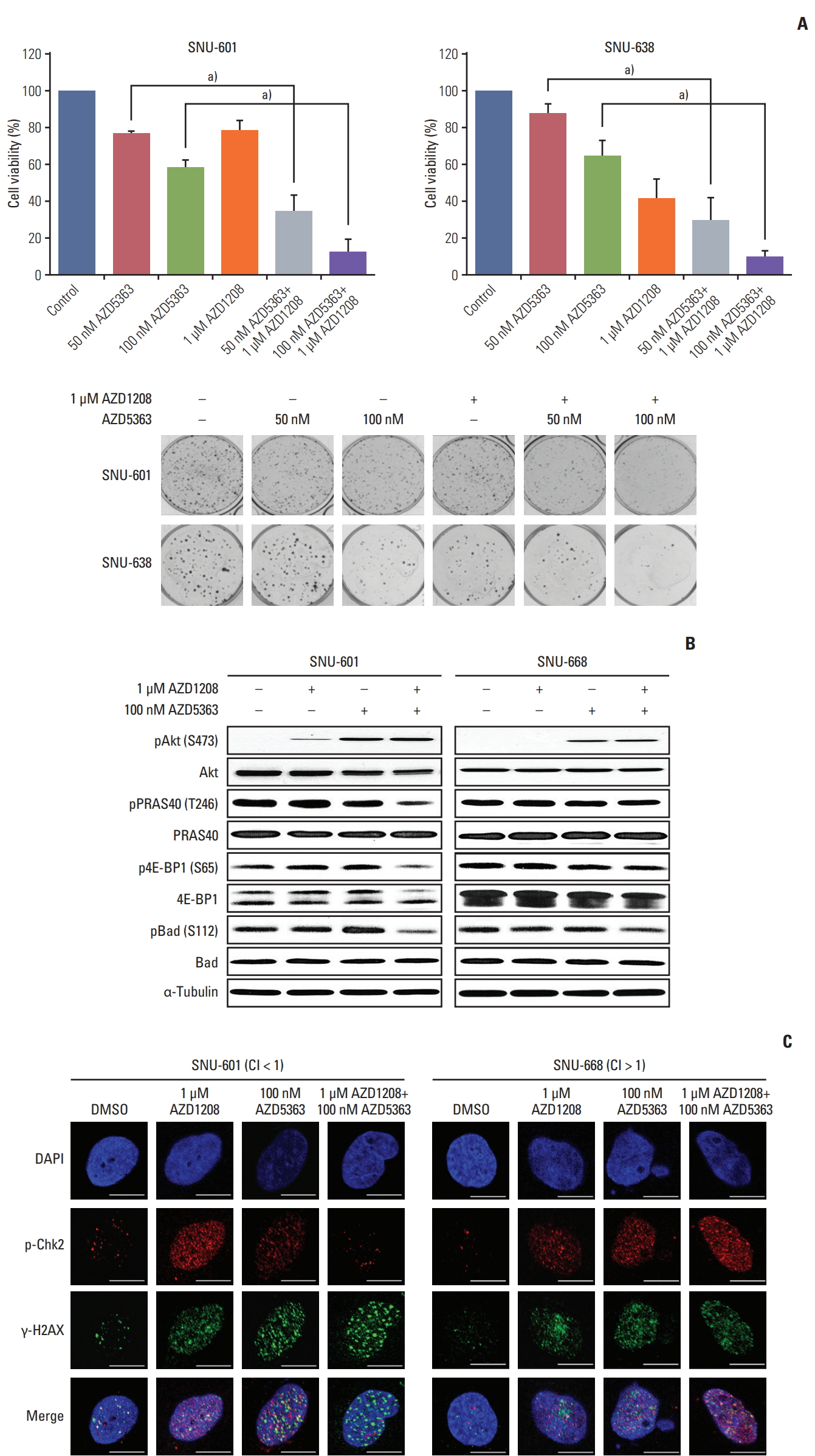
Overactivation of the Akt signaling pathway has been detected in gastric cancer [25]. Pim can induce resistance to Akt inhibition, and Akt modulates DDR signaling through interactions with DNA damage sensors, such as ATM, ataxia telangiectasia and Rad3-related protein (ATR), as well as DNA-dependent protein kinase catalytic subunit [26]. Thus, we hypothesized that co-administration of Akt and Pim inhibitors may exert more potent cytotoxic effects than treatment with either reagent alone because the combination could block the compensatory actions between Akt and Pim and disrupt the DDR pathway. We therefore monitored the combined effects of Pim and Akt inhibition using CFAs. As expected, the percentage of growth inhibition for the gastric cancer cell lines observed with dual treatment was significantly higher than for treatment with each reagent alone (S7 Table). In particular, the colony formation ability of AZD-1208-resistant SNU-601 cells was dramatically reduced by the combined treatment compared to exposure to AZD1208 alone (Fig. 6A).
To evaluate the signaling pathways involved in growth inhibition by combinatorial treatment with AZD1208 and Akt inhibitors, we examined the activities of 4E-BP1 and Bad, which are overlapping downstream molecules of the Pim and Akt cascades, respectively, in SNU-601 cells, in which synergistic effects were observed, and in SNU-668 cells, in which antagonistic effects were observed (Fig. 6B). We first confirmed the increased phosphorylation of Akt itself, which is consistent with observations reported in previous studies [18]. Combined administration of AZD1208 and Akt inhibitors markedly decreased phosphorylation of 4E-BP1 and Bad kinase in comparison to either agent alone. In addition, phosphorylation of PRAS40 was significantly reduced only in SNU-601 cells, resulting in down-regulation of mTOR signaling activity. Furthermore, we observed that co-administration of AZD1208 and AZD5363 led to a significant reduction of pChk2 expression and increased the number of γ-H2AX foci, a reasonable indicator of DNA double strand breaks, in SNU-601 cells (Fig. 6C). These results suggest that dual inhibition of Pim and Akt synergistically induce anticancer effects and could overcome resistance to AZD1208 through abrogation of DDR activity.
Discussion
Pim kinases are overexpressed in various types of tumors. Studies of the development of novel Pim kinase inhibitors have been published [27]. In a previous investigation, it was reported that AZD1208 is a potent pan-Pim kinase inhibitor with anti-tumor activity in acute myeloid leukemia [16], but the sensitivity of gastric cancer cell lines to Pim inhibition has not yet been assessed. In the present study, to our knowledge, we report the first assessment of the effects of AZD1208 and its underlying mechanisms of action in a gastric cancer cell line panel. AZD1208 down-regulated phosphorylation of Pim kinase substrates in a short period of time but did not induce apoptosis. Furthermore, AZD1208 reduced cell proliferation through autophagosome formation by LC3B in a long-term culture system. We also found that increased levels of DDR factors were associated with AZD1208 insensitivity. Moreover, we observed that the combination of AZD1208 with an Akt inhibitor had significant antitumor effects in gastric cancer cell lines, suggesting that while AZD1208 exerts therapeutic effects alone, these effects are more robust in combination with an Akt inhibitor.
Most studies of Pim inhibitors have shown that treatment with these reagents induces apoptosis through a reduction of phosphorylation of Pim kinase substrates [27]. However, we did not observe apoptotic cell death, although factors associated with cell survival, such as Bad and 4E-BP1, were down-regulated by Pim inhibition. On the other hand, it has been reported that the Pim inhibitor SGI-1776 induces limited apoptosis and autophagy in cases of multiple myeloma, although it was unclear how Pim inhibition caused autophagic cell death [28]. In our investigation, we determined that AZD1208 not only induced autophagic cell death but also attenuated DDR capacity. Wang et al. [21] noted that Pim-2 directly binds to and phosphorylates the cyclin-dependent kinase inhibitor p21 at Thr145. It is well-known that deregulated cell cycle progression causes replication stress and DNA damage [22]. Additionally, p53 induces autophagy in a damage-regulated autophagy modulator (DRAM)-dependent manner in the presence of DNA damage [29]. Based on the results of these studies, Pim inhibition by AZD-1208 led to the misregulation of intracellular signaling, resulting in increased DNA damage. Accumulation of DNA damage by Pim inhibition could also induce autophagy.
Activation of the ATM-Chk2 pathway was observed in SNU-601 cells following Pim inhibition. Interestingly, SNU-601 cells were found to have low levels of DRAM mRNA expression, which affected the initiation of autophagy (data not shown). These findings suggest that the DNA damage induced by AZD1208 is efficiently repaired, resulting in resistance to AZD1208.
Targeting the Akt signaling pathway has produced a limited antitumor effect to date due to drug resistance through a negative feedback loop as well as toxicity [14,30]. The present study is the first to report that inhibition of Pim could overcome these problems and induce cell death to a greater extent in gastric cancer cells. Moreover, the findings of the present study provide the first evidence that AZD1208 synergistically enhances AZD5363-mediated growth inhibitory effects as well as inhibits 4E-BP1 and Bad phosphorylation in most gastric cancer cell lines, which suggests that Pim inhibitors can act as sensitizers for Akt inhibitors in gastric cancer cell lines. Additionally, the synergistic effects of Pim and Akt co-targeting are attributable to decreased DDR capacity, which is a major mechanism of AZD1208 resistance, resulting in increased cellular sensitivity to AZD1208. These results provide a rationale for administering Pim and Akt inhibitors in combination to treat gastric cancer.
The findings in our study suggest that Pim inhibition, synergistically with Akt inhibition, can be a potential strategy for treating gastric cancer. Furthermore, our findings provide significant insights into the mechanism underlying the effects of Pim inhibition.