Introduction
In almost all cases, malignant rhabdoid tumor (MRT) is a rare and highly aggressive tumor characterized by its rhabdoid feature and biallelic loss of SMARCB1/INI1/hSNF5 [1]. For various reasons, MRT poses a challenge to clinicians. Despite the advancements in diagnostics tools, diagnostic challenges remain due to the myriad of anatomical locations of this tumor, the overlapping pathologic finding with other diseases, and the diverse immunophenotypic profile [2-8]. It has a predilection for infants and young children, peaking between birth and 3 years of age, an age group with risk of long-term sequelae following radiotherapy [1]. No standard treatment has been established for MRT, because systematic analysis of the outcome of the small numbers of heterogeneously treated patients has thus far been impossible based on the few retrospective reviews [1,9,10]. Most importantly, MRT is a highly aggressive tumor with published 5-year overall survival (OS) ranging between 15% and 36% [9].
We reviewed the clinical characteristics and treatment outcome of extra-cranial MRT patients treated in our institute. We thus aim to add to the current knowledge of this highly aggressive disease, and to aid in the development of better treatment strategies.
Materials and Methods
A retrospective medical record review was conducted on 11 children treated for extra-cranial MRT at Seoul National University Children’s Hospital between January 2003 and May 2013. All of the patients were pathologically confirmed as MRT by the institutional pathologist on the basis of morphological and immunohistochemical evaluations. They were all confirmed not to have concurrent involvements in the central nervous system with radiological imaging. Patients were staged according to the Surveillance, Epidemiology, and End Results (SEER) staging system [11].
Patients received multimodal therapies, including chemotherapy, surgical resection, radiotherapy, and high dose chemotherapy and autologous stem cell rescue (HDCT/ASCR). From November 2010, all extra-cranial MRT patients who achieved complete remission after conventional therapy underwent HDCT/ASCR with uniform conditioning with melphalan, etoposide, and carboplantin. This consisted of melphalan 140 mg/m2 on day –7, 70 mg/m2 on day –6, etoposide 200 mg/m2 and carboplatin 400 mg/m2 from days –8 to –5. Mobilized autologous peripheral blood stem cells were infused on day 0. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events ver. 4.03 (CTCAE v4.03).
Statistical analyses were performed using IBM SPSS ver. 19.0 (IBM Co., Armonk, NY). Kaplan-Meier method was used for analysis of OS and event-free survival (EFS), and log-rank test was used for subgroup comparisons. Statistical significance was defined as p < 0.05. The Institutional Review Board (IRB) at Seoul National University Hospital approved this retrospective medical record review (IRB No. H-1307-121-508).
Results
1. Clinical characteristics
Eleven patients (7 boys, 4 girls) were diagnosed with extracranial MRT during the 10 years and 5-month period in a single pediatric institute. Patients were diagnosed at very young ages; they presented at median age of 7 months old (range, 1 month to 13 years 1 month old) and were diagnosed at median age of 9 months old (range, 2 months old to 13 years 2 months old). Six patients (55%) were diagnosed during infancy and only one patient (9%) was diagnosed during adolescence (Table 1).
Primary sites of tumor were variable; six patients (55%) had renal MRT, all of which were unilateral, and five patients (45%) had soft tissue MRT, predominantly at deep axial locations such as submental, paraspinal, retrosternal, and coccygeal area. The most common presenting sign for renal MRT was gross hematuria (n=4), whereas that for soft tissue MRT was a mass lesion at primary sites (n=4) (Table 1).
Five patients (45%) had distant stage according to the SEER staging system; their most common site of metastasis was the lung (n=3) and all patients with lung metastases had multiple lung nodules. The rest of the patients had regional stage (n=3, 27%) or localized stage (n=3, 27%) (Table 1).
2. Diagnosis and loss of INI1 staining
Two of our patients were not pathologically confirmed as extra-cranial MRT on initial diagnosis. The primary tumor of patient No. 2 was too large for surgical excision and it had extensive perirenal hematoma with risk of severe bleeding which impeded incisional biopsy. It was suspected as a Wilms tumor based on the location of the primary tumor on radiologic imaging. After a cycle of chemotherapy with actinomycin and vincristine, the primary tumor was excised and was pathologically confirmed as renal MRT. Patient No. 5 was initially diagnosed as Wilms tumor with a needle biopsy specimen. He thus underwent chemotherapy with actinomycin and vincristine; however, despite chemotherapy, his condition deteriorated and he developed disseminated intravascular coagulation, metabolic acidosis, and oliguria. Thus, the initially biopsied specimen was further evaluated including INI1 staining and the diagnosis was revised to renal MRT due to the loss of INI1 staining (Table 1).
The tumor specimens of most of the patients had loss of INI1 staining. Specimens of 10 patients were studied for INI1 staining; those of nine patients (90%) had loss, and that of one patient (10%) had retained INI1 staining. Patient No. 3 was the only patient whose specimen had retained INI1 staining; he was also the only patient who showed unusual clinical characteristics, with late presentation during adolescence and an outstanding treatment outcome (Table 1).
3. Multimodal therapies
All patients (100%) underwent chemotherapy with various combinations of vincristine (n=10), cyclophosphamide (n=10), etoposide (n=10), doxorubicin (n=8), carboplatin (n=8), and ifosfamide (n=7) (Table 1).
Eight patients (73%) underwent surgical resection of the primary tumor; three patients (27%) underwent upfront surgery and five patients (45%) underwent delayed surgical resection after chemotherapy. Primary tumors were not resected in three patients (27%); tumor progressed in two patients and regressed in one patient (Table 1).
Six patients (55%) received local therapeutic radiotherapy. In four other patients (36%), therapeutic radiotherapy was not indicated; two patients had unresectable tumors which progressed and two patients had distant metastases. The remaining one patient, patient No. 3, did not undergo radiotherapy due to his extraordinary outcome; he is the only patient among survivors to not have received radiotherapy. Recipients of local therapeutic radiotherapy received median 27.0 Gy (range, 10.5 to 41.4 Gy) in median 16 fractions (range, 7 to 36 fractions), at median 40.2 months (range, 0.1 to 14.3 months) from diagnosis. The earlier three patients underwent radiotherapy postoperatively, but beyond November 2010, radiotherapy has been delayed until after HDCT/ASCR, and three patients underwent radiotherapy as such (Table 1).
From November 2010, patients who achieved complete remission with conventional therapy underwent HDCT/ASCR. Four patients (36%) have thus far undergone HDCT/ASCR with melphalan, etoposide, and carboplatin. They had median 8.6×108 mononuclear cells (MNC)/kg (range, 7.2×108 to 12.2×108 MNC/kg) and 8.4×106 CD34+ cells/kg (range, 3.3×106 to 24.1×106 CD34+ cells/kg) infused. Neutrophils engrafted on median day 10 (range, day 9 to day 10). During HDCT/ASCR, four patients had grade 3 febrile neutropenia according to CTCAE v4.03. One patient had additional grade 3 lung infection, grade 2 otitis media, and grade 2 sinusitis. Another patient had grade 3 acute kidney injury, and another patient had grade 1 acute kidney injury. All of these adverse events were transient.
4. Treatment outcome
Overall, three patients (27%) progressed during treatment and two patients (18%) relapsed after off-therapy. These five patients (45%) who progressed or relapsed, died of disease. There was no treatment related mortality (Table 1).
The three patients who progressed during treatment had initial lung metastases, which were multiple and which did not respond to initial chemotherapy. Thus, surgical resection of these multiple metastatic lung nodules was not feasible. These patients progressed at median 1.0 month (range, 0.9 to 1.7 months) from diagnosis, and died of disease at median 3.8 months (range, 2.3 to 4.5 months) from diagnosis (Table 1).
The sites of relapse were brain and lung. One patient relapsed in the brain, and another relapsed in the brain and the lung. Both of these patients were proven not to have concurrent disease of the central nervous system at initial radiological evaluation. They relapsed at median 3.7 months (range, 3.4 to 3.9 months) from off-therapy, and died of disease at median 4.3 months (range, 1.0 to 7.6 months) from diagnosis (Table 1).
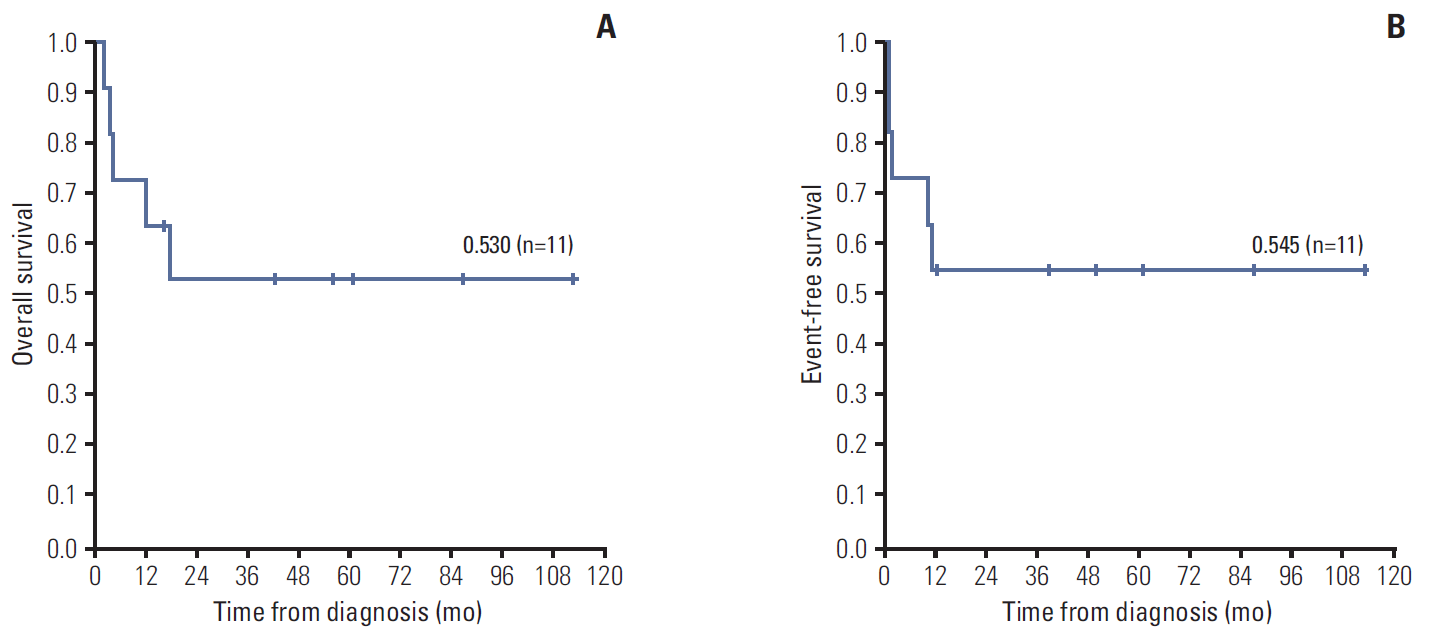
The OS of the total study population was 53.0% and their EFS was 54.5% with a median follow-up duration of 17.8 months (range, 2.3 to 112.3 months) (Fig. 1). The six survivors are currently disease-free for a median duration of 43.0 months from off-therapy (range, 17.5 to 104.7 months). In particular, patient Nos. 2, 3, and 6 are long-term survivors (Table 1).
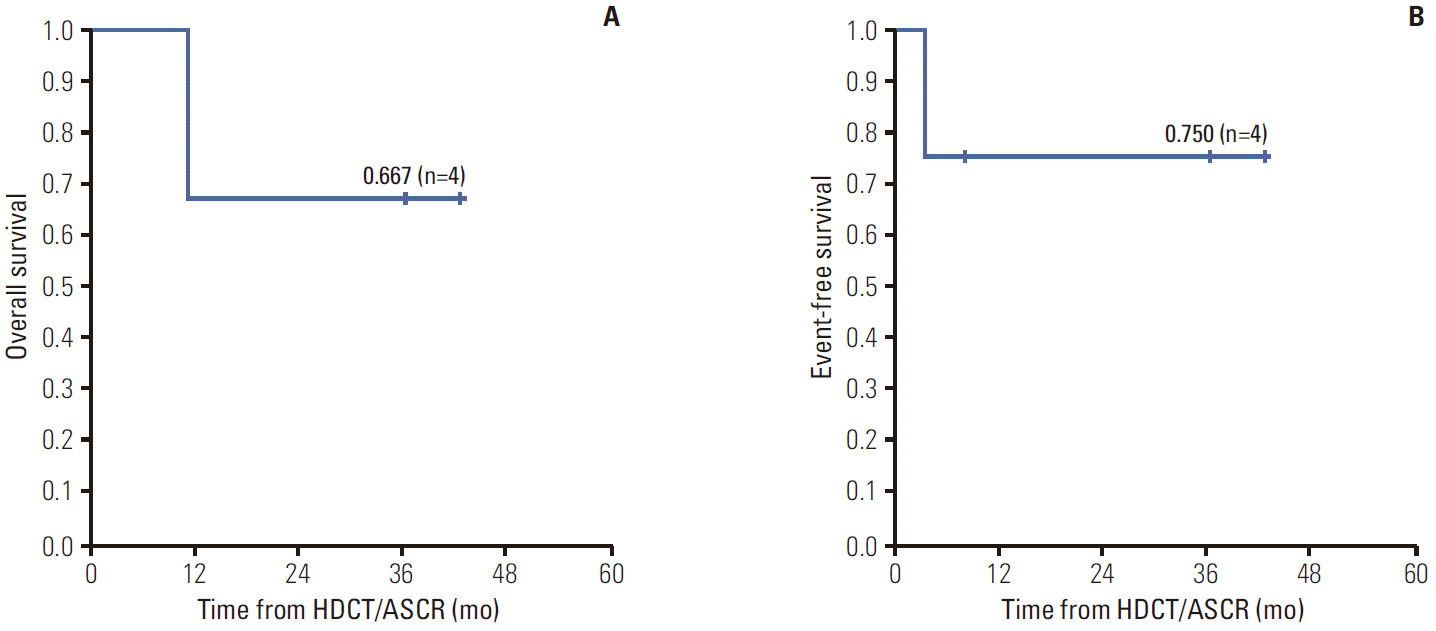
The OS of patients who underwent HDCT/ASCR was 66.7% and their EFS was 75.0% with a median follow-up duration of 23.8 months (range, 8.1 to 42.6 months) from HDCT/ASCR (Fig. 2). The three survivors are currently disease-free for a median duration of 36.3 months from off-therapy (range, 8.1 to 42.6 months).
In subgroup analysis, 75% of patients (6 of 8 patients) who were under 2 years old at diagnosis, died of disease whereas none of the three patients who were older at diagnosis, died of disease. In addition, 80% of patients (4 of 5 patients) with metastatic disease, died of disease whereas only 7% of those (1 of 6 patients) without metastasis, died of disease. However, worse survival of neither patients who were under 2 years of age at diagnosis nor patients with initial metastasis was supported with statistical significance on log-rank test.
Discussion
This is the first single institutional report on the clinical characteristics and outcome of extra-cranial MRT in Korea. In accordance with the significantly improved prognosis of MRT over the past decade, the current study showed promising results and some of the patients were long-term survivors [1,9-13].
Diagnosis of extra-cranial MRT as a distinct entity is a challenge; it is found in a myriad of anatomic sites, has overlapping pathological findings with other diseases, and shows histological heterogeneity with a diverse immunophenotypic profile [7]. The presence of rhabdoid feature is a histological hallmark of MRT but it is also observed in various other malignant tumors [6]. Thus with the discovery that the loss of INI1 gene contributes to the oncogenesis of MRT, INI1 antibody immunohistochemistry became an important tool in its diagnosis [7,8]. In the current study, the diagnosis of patient No. 5 was revised to extra-cranial MRT after additional INI1 staining. In fact, tumor specimens of most of our patients showed loss of INI1 staining. Nonetheless, there was a case of tumor specimen retaining INI1 staining despite pathologic confirmation as extra-cranial MRT based on morphological and other immunohistochemical studies. Previous literature has reported that genetic variations do exist in MRT, and that up to 20% have no alteration in the INI1 gene at the DNA or RNA level. Our patients who had retained INI1 staining also showed unusual clinical characteristics and extraordinary outcome. Thus, the possible impact of loss of INI1 gene on the clinical characteristics and prognosis of MRT needs further evaluation.
Extra-cranial MRT is treated with multimodal therapies, including chemotherapy, surgery, radiotherapy, and HDCT/ASCR, however, no standard treatment has been established to date. Anthracycline and actinomycine D have been shown to be important chemotherapeutic agents for MRT [13,14]. Also, alternating courses of the combination of vincristine, doxorubicin, and cyclophosphamide (VDCy) and the combination of ifosfamide, carboplatin, and etoposide (ICbE) have been suggested to be effective in metastatic MRT [15,16]. Currently, the treatment recommendation of “EU-RHAB” registry employs the use of doxorubicin, ifosfamide, carboplatinum, etoposide, vincristine, actinomycine D, and cyclophosphamide [1]. Our study population received combinations of chemotherapeutic agents including vincristine, cyclophosphamide, etoposide, doxorubicin, carboplatin, ifosfamide, and actinomycine D. However, patients included in the current study received heterogeneous combinations of chemotherapy regimens and thus an analysis on the benefits of a particular regimen could not be performed.
Complete surgical resection and radiotherapy have been reported to have survival benefits [9-11,13]. In the current study, 63% of the patients (5 of 8 patients) who managed to undergo complete surgical resections of the primary tumor as opposed to 33% of the patients (1 of 3 patients) whose initial masses were unresectable are currently alive with no evidence of disease. All of the survivors, except for one, have received therapeutic radiotherapy. However, due to the current small study population, the survival benefits of either surgery or therapeutic radiotherapy could not be analyzed with statistical significance.
HDCT/ASCR with etoposide, carboplatin, and melphalan (etoposide 200 mg/m2 on days –7 to –4, carboplatin 400 mg/m2 on days –7 to –4, melphalan 40 mg/m2 on days –3 to –2) have been shown to result in clinical responses and long-term survival in patients with poor prognostic factors [17]. The current study includes four patients who underwent HDCT/ASCR with melphalan, etoposide, and carboplatin. Despite the small number of patients, the OS of 66.7% and EFS of 75.0% with a median follow-up duration of 23.8 months (range, 8.1 to 42.6 months) are promising results. In addition, there were no treatment related mortality or severe adverse events. This suggests a possible role of HDCT/ASCR with melphalan, etoposide, and carboplatin in extra-cranial MRT.
Younger age has previously been found to be an independent risk factor, with patients either under 2 years of age or 3 years of age showing poorer survival [18]. In addition, association of metastatic disease at diagnosis with worse survival has been reported [10-12]. Although worse survival of neither the patients who were under 2 years of age at diagnosis nor the patients with initial metastasis was supported with statistical significance, there were tendencies of worse survival in these groups and further analysis is needed with a larger number of patients.
In accordance with some of the previous reports, lung was the most common site of metastasis in the current study [10,19]. In literature, complete resection of solitary lung nodules, whole-lung irradiation, and alternative courses of ICbE and VDCy chemotherapy has been suggested as possible strategies for lung metastases [14,15,20].
The current study is limited by its retrospective nature, the small cohort size, and the heterogeneous treatment strategies. A prospective, multicenter collaboration study on a larger number of patients treated with uniform treatment regimens will enable verification of the benefits of various treatment modalities, and identification of prognostic factors. This will thus aid in the development of better treatment strategies for this aggressive disease.
Conclusion
Extra-cranial MRT is a distinct disease entity and INI1 antibody immunohistochemistry may assist in its diagnosis. It is still a highly aggressive tumor in young children but the improved survival of our study population is promising. Multimodal treatment approach should be employed for this disease including chemotherapy, surgery, radiotherapy, and HDCT/ASCR. HDCT/ASCR with melphalan, etoposide, and carboplatin conditioning may be a promising treatment option for children with extra-cranial MRT.