Introduction
Histiocytes are cells fully differentiated from the monocyte/macrophage lineage and Langerhans/dendritic cells. Histiocytic and dendritic cell neoplasms are classified together by the World Health Organization, based on the cell function, rather than histogenesis. Histiocytic sarcomas are malignant proliferations of non-Langerhans histiocytes, excluding acute monocytic leukemias and associated neoplasms [1].
The pathogenesis of histiocytic sarcoma remains unclear. Furthermore, it can occur at any age, from infancy to adulthood, with several series reporting a greater predilection in males than in females. The most frequently involved organs are the skin, lymph nodes, and intestinal tract; however, it can also involve other organs. Clinical manifestations depend on the involved organs and can range from an asymptomatic single mass to systemic symptoms, including fever and weight loss.
To date, only tens of cases have been reported to involve the central nervous system (CNS) (Table 1) [2-14]. These patients usually have an extremely poor prognosis. We describe here a patient with primary CNS histiocytic sarcoma, with the involvement of multiple areas within the cerebral hemisphere and spinal cord, who was initially misdiagnosed as having demyelinating disease.
Case Report
A 59-year-old man was presented with progressive right side weakness and numbness in the left leg for 2 months. The deep tendon reflex on his right side was hyperactive, but there was no evidence of dysarthria, dysphagia, or defecation problems. His past history includes hypertension, diabetes mellitus, and coronary artery disease.
Laboratory findings were unremarkable. Blood glucose concentration was under control, and blood pressure was maintained around 140/90 mm Hg.
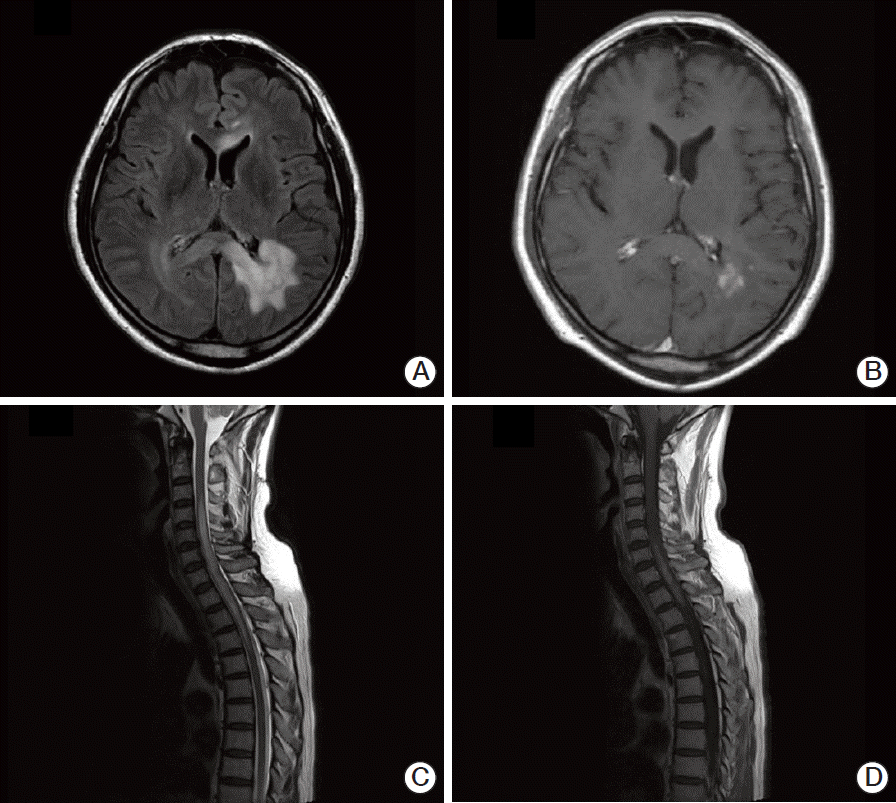
Magnetic resonance imaging (MRI) revealed multiple ill-defined T2 high signal intensity lesions in his brain and spinal cord. Brain lesions involved the left parieto-occipital periventricular area, extending to the right side through the splenium of corpus callosum, and both frontal lobes. These lesions showed high signal intensity on T2-weighted images, subtle increases in signal intensity on diffusion-weighted images, and patchy irregular enhancement following contrast injection (Fig. 1A and B). The lesions in the spinal cord had smooth margins and were distributed from the lower level of C3 to the upper level of T5, resulting in mild cord enlargement (Fig. 1C). T1-weighted enhanced sagittal image showed patchy enhancement in the corresponding spinal cord (Fig. 1D). Angiography showed no evidence of vascular abnormalities. The preliminary diagnosis was lymphoma or demyelinating disease.
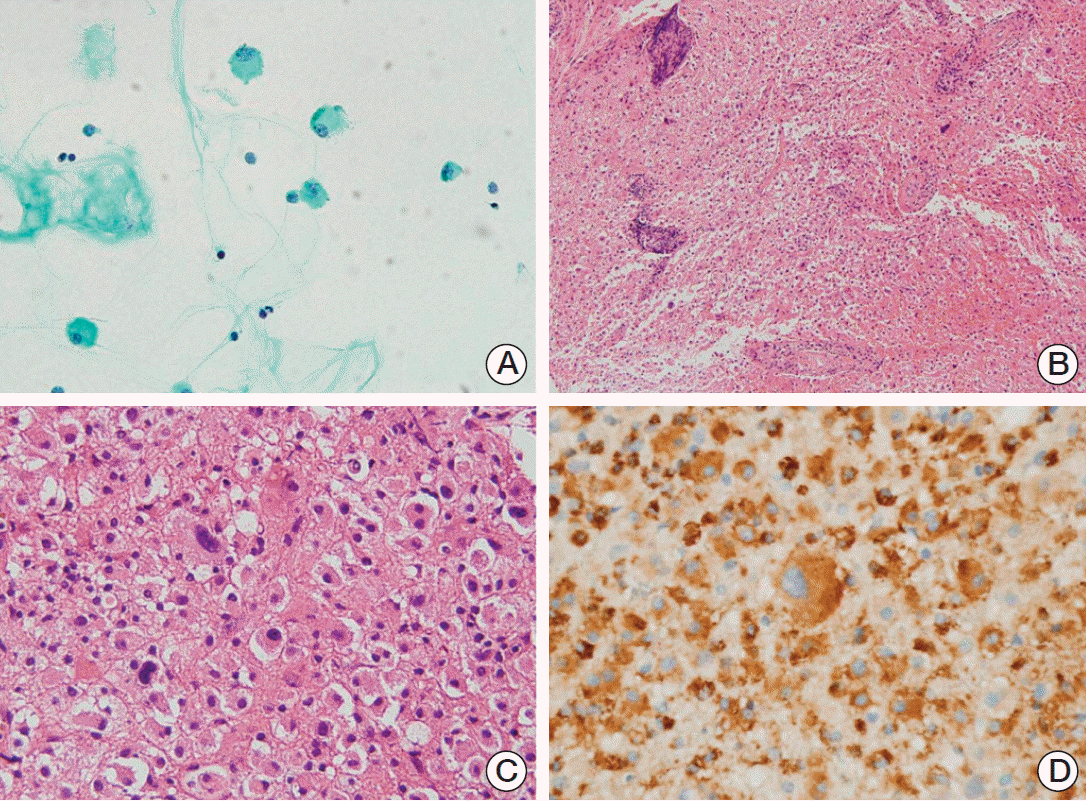
A sample of cerebrospinal fluid (CSF) was obtained for cytologic diagnosis. Giemsa staining revealed a few large cells intermixed with mature lymphocytes. The large cells had abundant cytoplasm and round to oval large nuclei with coarse chromatin, morphologically consistent with histiocytes (Fig. 2A).
Large cells with abundant cytoplasm, similar to the cells in CSF smear, were mostly present in a stereotactic biopsy of the lesion in the left parieto-occipital area (Fig. 2B). Morphologically, these cells had characteristics of histiocytes, a finding supported by their positivity for CD68. Blood vessels within the lesion were surrounded by mature, normal-looking lymphocytes. Although a few bizarre giant cells were present (Fig. 2C), they appeared similar to adjacent histiocytes, except for their size, and were positive for CD68 (Fig. 2D). Due to the abundance of histiocyte infiltration in the absence of necrosis, hemorrhage, and neutrophil infiltration, the lesions were considered to have resulted from demyelinating disease.
Based on a clinical and pathologic diagnosis of tumefactive multiple sclerosis, the patient was treated with steroid pulse therapy and plasmapheresis, and subsequently with azathioprine. However, his neurologic symptoms did not improve. A follow-up MRI, performed 3 months after the first biopsy, showed that the extent of the left paraventricular lesion had widened, and that there were new lesions in the T4-T9 and T10-T11 levels of the spinal cord.
The possibility of neoplasm was considered likely, and a second biopsy was performed at the deeper part of the adjacent lesion from the first site. Three months have passed since the first biopsy. The histologic features of the second biopsy specimen were generally the same as that of the first, showing large cells with abundant eosinophilic to amphophilic cytoplasm (Fig. 3A). The nuclei were located eccentrically and chromatin was coarsely clumped with occasional visible nucleoli. The nucleus size was larger, and nuclear atypia was more severe than in normal histiocytes. Bizarre cells, with very large hyperchromatic nuclei, were more frequent than in the first biopsy specimen. Immunohistochemistry showed that the cells, including the large bizarre cells, were positive for CD68, CD163, and S-100 proteins, which suggest their histiocytic nature. The cells were negative for glial fibrillary acid protein (GFAP) and cytomegalovirus (CMV) antigen, excluding the possibilities of glioblastoma and CMV infection, respectively (Fig. 3B-D). The diagnosis was revised to histiocytic sarcoma.
For a staging evaluation, chest and abdominal computed tomography, positron emission tomography, and bone marrow studies were performed, and there was no evidence of lymphoma involvement other than the brain and spinal cord.
The patient was treated with two courses of high dose methotrexate (MTX) as the first line of treatment. A total dose of 3,500 mg/m2 MTX was administered for 120 minutes on day 1, every 2 weeks. Leucovorin rescue was followed 24 hours after MTX administration. As the tumor did not respond to two courses of the treatment, the regimen was changed to cytarabine (Ara-C) in a dose of 3,000 mg/m2 on day 1 and 2, every 4 weeks. MRI and positron emission tomography after two courses of Ara-C showed increased tumor extent and linear enhancement of the anterior aspect of the spinal cord at the lumbar spine level, indicating disease progression. The patient died 2 months after the second biopsy, and 8 months after the onset of symptoms.
Discussion
Since the first description of histiocytic sarcoma in 1970, a few cases with CNS involvement have been reported. Patients can present symptoms thatrange from a simple headache to neurologic deficits. Neurologic deficits can significantly impair a patient’s general condition, making the disease harder to manage. Histiocytic sarcomas can involve any site within the CNS, including the brain parenchyma, spinal cord, and meninges. Our patient was apparently the third to show involvement of both the brain and the intramedullary region at presentation [3,14].
A histologic diagnosis of histiocytic sarcoma requires the presence of cells with morphologic characteristics of histiocytes and an appropriate immunophenotype [1]. Morphologically, these tumors are composed of highly cellular non-cohesive proliferations of round, oval or polygonal cells with indistinct cell membranes. The cytoplasm is abundant and eosinophilic to amphophilic. The nuclei vary in size and shape, from small and round to large and irregular, which can sometimes even exhibit a lobulated shape. Very large bizarre and occasionally multinucleated cells may be observed. The tumor cells are positive for histiocyte-associated antigens, including CD68, lysozyme, HAM-56, 1-antitrypsin, α1-antichymotrypsin, and nonspecific esterase, but are negative for B cell, T cell, myeloid, follicular dendritic cell, and Langerhans cell markers (CD1a), as well as CD30.
The patient was first diagnosed with a demyelinating disease. The lesions were multifocal, involving the periventricular areas and spinal cord, and a biopsy revealed many histiocytes with inflammatory infiltrates. The small biopsy specimen contained mostly monomorphic histiocytes, with only a few large cells that were overlooked. Histiocytic sarcoma should be distinguished from demyelinating diseases, which are treated with steroids. Both diseases are characterized by abundant histiocytic infiltrates, and their differentiation is further complicated when neoplastic histiocytes are intermixed with prominent inflammatory components [9,10]. The tumor cells of histiocytic sarcoma vary in size, from 18 to 26 μm, with many larger than non-neoplastic histiocytes [1,9], and with abundant cytoplasm and large hyperchromatic nuclei. Nuclear atypia and pleomorphism with occasional large bizarre nuclei are the key features that distinguish histiocytic sarcoma from non-neoplastic histiocytic infiltration in demyelinating diseases.
Histiocytic sarcoma can be confused with other histiocytic or histiocyte-like lesions in the brain, such as Langerhans cell histiocytosis, Rosai-Dorfman disease, and juvenile xanthogranuloma [1,15]. Histiocytes in Langerhans cell histiocytosis have indented or lobulated nuclei without significant atypia, which are positive for CD1a, and are accompanied by eosinophilic infiltration. Rosai-Dorfman disease is characterized by emperipolesis and infiltration by numerous plasma cells, with histiocytes having voluminous, amphophilic cytoplasm, and round nuclei with prominent nucleoli. In juvenile xanthogranuloma, the histiocytes are often lipidized, with a few showing significant atypia [15].
Primary CNS tumors with pleomorphic or large bizarre cells can mimic histiocytic sarcoma [1]. Glioblastoma, the most common primary intraparenchymal tumor of the CNS, often has many bizarre and atypical large cells. However, these cells possess cytoplasmic processes, resulting in a fibrillary appearance in the background, which are positive for GFAP, and are negative for histiocytic markers. Pleomorphic xanthoastrocytoma is a prototype of tumors with pleoPrimary CNS tumors with pleomorphic or large bizarre cells can mimic histiocytic sarcoma [1]. Glioblastoma, the most common primary intraparenchymal tumor of the CNS, often has many bizarre and atypical large cells. However, these cells possess cytoplasmic processes, resulting in a fibrillary appearance in the background, which are positive for GFAP, and are negative for histiocytic markers. Pleomorphic xanthoastrocytoma is a prototype of tumors with pleomorphic large cells; but spindle cells, lipidized cells, and eosinophilic granular bodies are present, in addition to large bizarre cells. These tumors can be differentiated from histiocytic sarcoma by positivity for GFAP and synaptophysin, and negativity for histiocytic markers. Anaplastic large cell lymphoma may be histologically indistinguishable from histiocytic sarcoma, due to high cellularity and significant nuclear atypia with occasional large bizarre cells. Anaplastic large cell lymphomas, however, are positive for CD30, but negative for histiocytic markers present in histiocytic sarcoma [1,9].
Treatment strategies for histiocytic sarcoma include surgical resection, steroid treatment, chemotherapy, and radiation. Although most tumors are unresectable due to location and number, a total excision, if feasible, should be considered since it may provide a better prognosis, with some patients surviving over 1 year from a diagnosis after complete resection [4,10].
Steroids may relieve the symptoms temporarily by reducing peritumoral vasogenic edema and oncolytic effect. However, steroid treatment should be avoided before diagnosis, as the oncolytic effects of steroids may interfere with accurate diagnosis.
The permeability of the blood-brain barrier (BBB) should be considered prior to prescribing chemotherapy. A high dose of MTX and Ara-C, which can easily penetrate the BBB, are the main agents used in the treatment for primary CNS lymphoma, which improve the overall survival. Both of these agents, however, were ineffective in our patient. Whole-brain irradiation may improve the responses to chemotherapy, albeit the rate of neurotoxicity is high.