Tassi and Wellstein: Tumor Angiogenesis: Initiation and Targeting - Therapeutic Targeting of an FGF-Binding Protein, an Angiogenic Switch Molecule, and Indicator of Early Stages of Gastrointestinal Adenocarcinomas -
Tumor Angiogenesis: Initiation and Targeting - Therapeutic Targeting of an FGF-Binding Protein, an Angiogenic Switch Molecule, and Indicator of Early Stages of Gastrointestinal Adenocarcinomas -
Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC, USA.
Correspondence: Anton Wellstein, Georgetown University, Lombardi Cancer Center, Research Building E311, 3970 Reservoir Rd. NW, Washington, DC 20057. (Tel) 202-687-4771, (Fax) 202-687-4821, wellstea@georgetown.edu
Tumor angiogenesis has been related to the initiation as well as progression toward more aggressive behavior of human tumors. In particular, the activity of angiogenic factors is crucial for tumor progression. We previously characterized a secreted fibroblast growth factor-binding protein (FGF-BP) as a chaperone molecule, which binds to various FGFs, enhances FGF-mediated biochemical and biologic events and importantly is a crucial rate-limiting factor for tumor-dependent angiogenesis. We generated monoclonal antibodies that target FGF-BP protein and used them as a tool to evaluate frequency and pattern of FGF-BP expression during the malignant progression of pancreas and colorectal carcinoma in archival tissue samples. We found that FGF-BP is dramatically upregulated during the initiation of colorectal and pancreatic adenocarcinoma. Crucial genetic events underlying the initiation and progression of colorectal and pancreatic adenocarcinoma with a particular focus on the modulation of angiogenesis and antiangiogenic therapies are discussed. We propose that the upregulation of the secreted FGF-BP protein during early phases of pancreas and colon cancer could make this protein a possible serum marker indicating the presence of high-risk premalignant lesions. Furthermore, the biological activity of FGF-BP is neutralized by monoclonal antibodies suggesting the potential for antibody-based therapeutic targeting.
Classically, angiogenesis is a process of new blood vessel formation from preexisting vessels (1). More specifically, angiogenesis is the mechanism mediating the growth and modification of a capillary network (2,3). Although normal endothelium is generally quiescent in the adult organism, angiogenesis is prominent during conditions of tissue repair and remodeling (eg, menstrual cycle and mammary gland involution), and pathologic events, such as wound healing, rheumatic diseases, diabetic retinopathy, inflammation, and malignancy (2,4).
Indeed, angiogenesis is an essential process in neoplastic growth. In fact, for a tumor to develop a highly malignant and deadly phenotype, it must first recruit and sustain its own blood supply (5). Since cells cannot survive if they lack an adequate supply of oxygen and nutrients, the expansion of a tumor mass beyond 1 mm in diameter is therefore dependent on the recruitment of its own vascular supply, by angiogenesis and/or blood vessel cooption (5~7). The process of angiogenesis is also sustained by secretion of specific angiogenic factors by cancer and non-cancer cells. Angiogenic factors can initiate a multi-step process that begins with vasodilatation, followed by the enhancement of vessel permeability and stroma degradation. Angiogenic factors can also promote endothelial cell migration and proliferation. As a result, endothelial cells converge and tightly assemble to generate a new lumen or they intercalate with endothelial cells in a preexisting vessel to give rise to capillary elongation. The newly formed capillary-like vessels undergo final remodeling, in which they rearrange from an irregular into a structured plexus of branching vessels and capillary loops (8). A hallmark of solid tumors is represented by abnormal vasculature. Tumor vessels are organized in a chaotic fashion, characterized by sinuous, extended, and enlarged vessels and are randomly fused with either arterioles or venules, creating an atypical microcirculation (9). In addition, tumor vessels are leaky, as a result of endothelial cell aberrant lining, fenestrations, vesicles and transcellular holes, widened inter-endothelial junctions, and a discontinuous or absent basement membrane (9~12). Tumor vessels may also lack functional perivascular cells (13) and their walls can be formed by a mosaic of alternating endothelial and cancer cells (vasculogenic mimicry), thereby exposing cancer cells to the lumen (14,15). As a result, neoplastic regions are often hypoxic and acidic due to the chaotic and slow blood flow (2,16~19). Tumor vessels infiltrate the neoplastic mass, sustaining its growth and its metastatic perfusion.
The abnormal feature of the tumor vasculature is likely to represent an imbalanced expression of angiogenic factors and inhibitors within a tumor. The blood vessel growth in normal tissues is regulated by the action of angiogenic stimulators, "proangiogenic factors" and angiogenic inhibitors, "antiangiogenic factors". The stimulators include: basic fibroblast growth factor (FGF-2), a potent mitogen for endothelium, VEGF, the most potent endothelial chemoattractant; cyclooxygenase 2 (COX-2), matrix metalloproteinases (MMPs), and transforming growth factor-beta 1 (TGF-beta1). Antiangiogenic factors counteract the effect of the angiogenic stimulators and therefore keep the normal blood vessels growth under a strict and well balanced control. These antiangiogenic factors include tissue inhibitor of metalloproteinase 1 (TIMP-1), interleukin-10 (IL-10), angiostatin, endostatin, and interferon (IFN) (20). Thus, activation of the angiogenic switch during early stages of tumor development suggests that regulation of the angiogenic process is likely to be a rate-limiting step in the progression from small lesion to extensive disease (3,5,21).
FIBROBLAST GROWTH FACTORS AND FIBROBLAST GROWTH FACTOR-BINDING PROTEIN
The FGF family comprises to date more than 20 distinct, structurally-related proteins that exert biologic effects in different cells and organ systems, including tumor growth and angiogenesis (22~25). FGFs are heparin-binding proteins, which interact with low affinity heparan sulfate proteoglycans (HS-PGs). HSPGs can protect FGFs from thermal denaturation and proteolysis and can increase FGF receptor affinity and facilitate FGF binding to cell surface receptor. In addition, ECM-associated HSPGs modulate FGF bioavailability by generating a local reservoir for the growth factor and allowing a sustained stimulation of endothelial cells (22,24). One mechanism that explains how these FGFs are mobilized from the extracellular storage includes the action of heparinases or other glycosaminoglycan-degrading enzymes (22,24). An independent mechanism is the action of a secreted FGF binding protein (FGF-BP) that functions as an extracellular chaperone for the locally-stored FGFs. Interestingly, FGF-BP is likely to complement the role of heparan sulfate in mediating FGF-dependent signaling and mitogenesis (26~28). FGF-BP was first described in 1991 by Wu et al as a low-affinity heparin binding protein, isolated from A431 human epidermoid carcinoma cells (29). FGF-BP binds to FGF-1 and FGF-2 in a noncovalent, reversible manner facilitating the release of the growth factor from the ECM and presenting it to FGF receptors (27,30~32). Numerous studies conducted in our laboratory have confirmed a role for FGF-BP as an extracellular chaperone for FGFs that enhances FGF-dependent biochemical and biologic functions. In murine NIH-3T3 fibroblasts as well as in bovine GM7373 endothelial cells, FGF-BP was shown to enhance FGF-2-induced MAPK signaling pathway and cell proliferation and we showed recently that the interaction domain with FGF-2 is a short domain in the C-terminus of the FGF-BP protein (26,27,33). Furthermore, FGF-BP was responsible for enhancing FGF-2-mediated angiogenesis in a chick chorioallantoic membrane (CAM) assay (27). Overexpression of FGF-BP in a chicken transgenic model resulted in embryonic lethality due to massive disruption of blood vessel structure and integrity and subsequent hemorrhage (34).
FGF-BP is expressed below the level of detection by Northern blotting in normal adult human tissues. However, its expression is considerably elevated in various tumors, including skin, head and neck, cervical, lung, squamous cell carcinomas, breast, colon, and pancreatic adenocarcinomas (30,31,35~37). Expression in FGF-BP-negative cell lines indicated FGF-BP as a rate- limiting factor for tumor growth and angiogenesis in a nude mouse model (30). Similarly, ribozyme-targeting and depletion of endogenous FGF-BP from human squamous cell carcinoma and colorectal cancer cell lines resulted in a significant reduction of tumor growth and angiogenesis. These findings support a potential role of FGF-BP as an angiogenic switch in human neoplasia (31).
ANGIOGENESIS IN COLORECTAL AND PANCREATIC ADENOCARCINOMAS
1) Colorectal cancer
Colorectal cancer is the third most common type of cancer among men and women in the United States. Over 95% of colorectal cancers are adenocarcinomas. The other 5% is represented by other, more rare, types of colorectal neoplasias. Eighty-five percent of colorectal cancer patients undergo surgical removal of the primary tumor. Although early stages of the disease (Duke's A and B) are linked with a good post-operative prognosis and 80~95% of remission rate, tumor cell infiltration through the serosa (B2 stage) and lymph node metastasis (C stage) reduce the 5-year survival to 25% to 60% (38). The American Cancer Society estimates that there will be approximately 148,000 new cases of colorectal cancer and 55,000 deaths in the 2006 in the United States. Risk factors include age, a diet rich in fat and cholesterol, ethnic background, alcohol assumption, inflammatory bowel disease (ulcerative colitis, Crohn's disease), and family history, including hereditary polyposis and nonpolyposis syndromes. Reductions in morbidity and mortality can be achieved through detection and treatment of early-stage colorectal cancers and the identification and removal of adenomatous polyps. Colorectal cancer screening tests, such as fecal occult blood test (FOBT), fecal himmunochemical test (FIT), flexible sigmoidoscopy, and colonoscopy, performed in asymptomatic 50 or more year old individuals, allows accurate detection of early stage cancerous and precancerous lesions.
Pioneering research by Vogelstein and colleagues conducted in the past 15 years has significantly contributed in the understanding of the molecular basis of colorectal cancer initiation and progression (39). Unlike other types of malignancies, colon adenocarcinomas develop through a progression of clinical and histopathologic processes that proceed from a single crypt lesion through small benign tumors (adenomatous polyps) to malignant and invasive carcinomas (40) (Fig. 1). The earliest genetic alteration promoting the initiation of colorectal malignant transformation is a germline mutation in the adenomatous polyposis coli (APC) tumor suppressor gene (39,41,42) that generates a truncated APC protein unable to interact and suppress beta-catenin activity (43~49). The resulting sustained beta-catenin overexpression promotes the activation of growth-promoting oncogenes, such as cyclin D1 or c-myc (50~52). Interestingly, mice carrying an endogenous APC mutation (ApcMin/+) similar to that found in human colorectal cancers do not survive beyond 4 months of age as a result of development of adenomatous polyps that rapidly progress to colorectal malignancies (53). Mutation of the ras proto-oncogene has been detected in various human cancers, including 95% of pancreatic cancers and up to 50% of large bowel adenocarcinomas (54~56). Since this mutation has been identified in both small and large colon adenomas and also in adenocarcinomas (57~59), it is likely to represent a crucial step contributing to the transition from intermediate to late adenoma or adenocarcinoma (60). Chromosome arm 18q deletions, resulting in the mutation and reduced expression of DCC (deleted in colorectal carcinoma) tumor suppressor gene (61~64) and SMAD4/DPC4 (deleted in pancreatic carcinoma 4) (65~67) accounts for a later event associated with colon cancer progression through the stages of late adenoma to carcinoma (68). Loss or inactivation of p53 tumor suppressor gene, reported in a high percentage of colorectal cancers, is likely to be the latest event during disease progression (69). Disruption of p53 by gene targeting in human colon cancer cells results in cell resistance to different chemotherapeutic agents (70). Therefore, loss of p53 in human colorectal cancers may account for the inefficacy of chemotherapy and decreased patient survival (71~74).
In colorectal cancer, several studies indicate angiogenesis as a crucial event leading to colon cancer progression. As a matter of fact, colorectal cancer is one of the best-studied models of tumor angiogenesis (91). As in many other tumors, several angiogenic regulators have been recognized in colon cancer, including VEGF, PDGF, thrombospondin, and angiopoietins (91,92). Likewise, overexpression of FGF and FGFRs in colon cancer cells and tissues, as well as increase of FGF-2 serum levels in patients with advanced colon cancer, have been extensively reported (93~98).
2) Pancreatic cancer
Pancreatic cancer is a cause of death of about 30,000 individuals each year in the Unites States (75). Although pancreatic cancer is recorded in only 2% of new cancer patients, it is the fifth leading cause of cancer-related death. Due to the lack of an efficacious early diagnostic test and to the manifestation of symptoms during late-stage disease, the malignancy is generally diagnosed after invasion and metastasis in surrounding tissues disabling patients to undergo curative resections. Another element playing a role in poor prognosis is the pancreatic cancer cell resistance to cytotoxic agents and radiation (76,77). Pancreatic adenocarcinoma develops through a step-wise progression from distinct epithelial lesions in the small interlobular ducts, namely pancreatic intraepithelial neoplasias (PanINs). PanINs can be flat (PanIN-1A), papillary without atypia (PanIN-1B), papillary with atypia (PanIN-2), or with characteristics of carcinoma in situ (PanIN-3) (78) (see Fig. 2). The molecular genetics of pancreatic adenocarcinoma have been well studied. Of these tumors, 80~95% have mutations in the K-ras gene (79,80), and 85~98% have mutations, deletions, or hypermethylation in the p16 tumor suppressor gene. Of these cancers, 50% have mutations in p53 and about 55% have homozygous deletions or mutations of DPC4/Smad4 and BRCA2. Some of these mutations can also be found in high-risk precursors of pancreatic cancer. For example, in chronic pancreatitis, 30% of patients have detectable mutations in p16 and 10% have K-ras mutations (81).
Although pancreatic cancer is not a grossly vascular tumor, it is often characterized by enhancement of tumor-dependent angiogenesis (82). A growing line of evidence has shown that various FGFs, such as FGF-1, FGF-2, FGF-5, FGF-7 (83~86) and FGF receptors (87~90) are upregulated in pancreatic cancer tissue samples and cell lines. These findings suggest that FGF-dependent downstream biologic events are likely to play an important role in the pathobiology of pancreatic cancer.
FIBROBLAST GROWTH FACTOR-BINDING PROTEIN IS AN EARLY MARKER IN GASTROINTESTINAL CANCERS
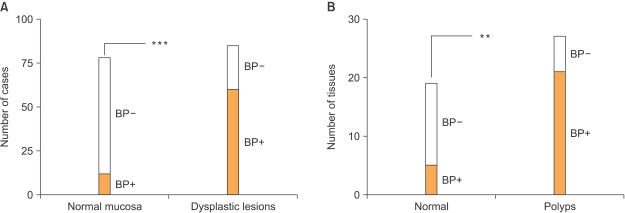
Our laboratory has recently focused on the analysis of FGF-BP expression and regulation during the progression of gastrointestinal cancers. In an initial study by Ray and colleagues (36), immunohistochemical analysis of normal and pathologic human colon biopsies using a polyclonal anti-FGF-BP antibody showed a significant upregulation of FGF-BP in dysplastic lesions, originating at the level of single crypts. Upregulation of FGF-BP protein was detected in a small portion (16%) of 76 apparently normal colon samples. However, expression of FGF-BP was detected in most samples with moderate to severe dysplasia (62 of 85; p<0.0001 normal versus dysplastic mucosa) (Fig. 3A). Furthermore, coincident with FGF-BP overexpression in dysplastic crypts, an increase of blood vessel density was found by immunostaining of the lamina propria with a CD31 antibody (from 80±7 to 154±9 vessels/field). The fact that specimens from different inflammatory bowel diseases, such as ulcerative colitis or Crohn's disease, did not show a significant FGF-BP upregulation prompted the authors to link FGF-BP overexpression in the onset of colon cancer to an early genetic occurrence. Analysis of FGF-BP mRNA by ISH in APCMin/+ mouse colon specimens showed a significant upregulation of FGF-BP in colon adenomas (21 of 27, >30% of the adenoma surface area). In contrast, little if any FGF-BP expression was noted in adjacent normal intestine (5 of 19; p<0.001, normal versus adenomas) (Fig. 3A). Interestingly, a prominent correlation between FGF-BP upregulation and cytoplasmic and nuclear beta-catenin staining in dysplastic colon was seen but not in a dextran sulfate-induced model of inflammatory colon disease (36). This finding further corroborated the hypothesis that FGF-BP is likely to be an early response gene during the initiation of colon cancer. Moreover, based on these findings and promoter-reporter studies, FGF-BP was shown to be a direct target of the activation of the Wnt/beta-catenin pathway (Fig. 1) and therefore represents an early event for colon cancer initiation.
Due to the potential significance of this protein in different human cancers, we generated monoclonal antibodies (mAbs) to FGF-BP and employ them to identify of FGF-BP protein in various bioassays, such as ELISA, western blot, immunofluorescence (Fig. 4) and himmunohistochemistry (Fig. 5) (37). Monoclonal antibodies were applied for immunohistochemical (IHC) detection of FGF-BP in tissue microarrays containing a series of tissue samples from different stages of both colon and pancreatic cancers. In parallel, detection of FGF-BP mRNA in the same tissue slides was performed by in situ hybridization. High expression levels of FGF-BP protein and mRNA were found in adenomatous polyps (Fig. 5) and the upregulation was sustained throughout all other stages of colorectal cancer progression, such as invasive adenocarcinoma and metastases, relative to the levels detected in normal specimens (p<0.0001). In addition, FGF-BP protein and mRNA were found dramatically upregulated during the early stages of pancreatic cancer progression, such as low-grade (PanIN1 and 2) and high-grade PanIN as well as invasive adenocarcinoma. Positive FGF-BP expression was also evidenced in some pancreatitis specimens, whereas very low or no expression was detected in normal pancreatic ducts. Nonadenocarcinoma samples showed expression of FGF-BP in 23% of samples at low levels, although this expression still represented a significant increase relative to normal specimens (p=0.0044). A highly significant correlation resulted from comparison between protein and mRNA expression in all samples analyzed (37).
Based on these observations, it is tempting to speculate that FGF-BP is an early regulatory gene, activated during the early phases of colon and pancreatic cancer progression. It is also possible that, as a consequence of FGF-BP overexpression in the early stages of colon and pancreatic cancers, an enhancement of tumor-dependent angiogenesis and invasion may occur, due to the increase of FGF-BP-mediated FGF bioavailability.
Although the primary purpose of generating mAbs to FGF-BP was to generate better detection tools, we sought to also assess the potential of any of these mAbs to inhibit the function of FGF-BP in a bioassay. For this we used a previously established in vitro model (30) and identified the one monoclonal Antibody (mAb) as a blocking antibody that reduces anchorage- independent growth dependent on FGF-BP (37) (Fig. 6). This suggests these antibodies could be used to therapeutically target FGF-BP in cancer patients.
ANTIANGIOGENIC THERAPIES
Anti-angiogenic inhibitors are used as part of therapeutic strategies addressed to arrest endothelial cell proliferation and differentiation in the tumor environment, thereby preventing cancer growth and metastasis. In tumor masses, one capillary vessel with a single endothelial cell is able to provide nutrients to approximately 100 cancer cells. For this reason, inhibition of endothelial cell growth might represent an amplification factor in halting the progression of tumor growth. In addition, since physiological angiogenesis is downregulated in adults, minimal side effects are obtained upon prolonged anti-angiogenic treatments (100,101). Neutralization of pro-angiogenic factors expression or activity, inhibition of the capillary basement membrane turnover, and inhibition of endothelial cell proliferation, migration, and differentiation are the diverse effects that the therapeutic application of specific anti-angiogenic agents can target.
New antiangiogenesis therapies are emerging. In February 2004, the United States Food and Drug Administration (US-FDA) approved Bevacizumab (Avastin), the first angiogenesis drug for first line treatment of patients with metastatic colorectal cancer. Avastin is a humanized variant of monoclonal antibody (93% human, 7% mouse) that binds to VEGF with high affinity and neutralizes all VEGF-A isoforms, thereby blocking VEGF receptor binding. Due to the abnormal features of tumor vessels, angiogenic inhibitors should normalized these tumor vessels, in order to achieve better cytostatic drug delivery within the tumor tissue (102). For instance, prevention of vascular leakage would decrease the interstitial pressure within the tumor mass and increase blood perfusion and chemotherapeutic drug delivery when antiangiogenesis drugs and chemotherapeutic agents are combined. Indeed, the administration of Avastin with chemotherapeutic drugs has resulted in a significant improvement of median survival, free survival, percentage of response rate and median duration of response (103,104). Analogous clinical benefits were obtained in patients affected by non-squamous non-small cell lung cancer (NSCLC), breast and renal cancer when treated with a combination of Avastin and chemotherapy (105). It is then possible that the antitumor activity exerted by VEGF antagonists in combination with cytostatic agents results in an ameliorated VEGF-induced leakage and interstitial pressure and therefore in a more efficient cytostatic drug penetration (106).
CONCLUSION
Early events that initiate the stepwise progression of premalignant lesions of colon epithelia and pancreatic ducts to invasive cancer may provide circulating markers for the detection of such lesions. FGF-BP, as a secreted protein, could be such a marker to identify subjects at an increased risk for developing invasive cancers. Therefore, generation of a sensitive ELISA assay able to detect FGF-BP in patient sera may represent an important diagnostic screening method for early detection of colon and pancreatic premalignant lesions, thereby allowing early medical intervention.
Notes
This work was supported in parts by grants of the NIH/NCI R01 CA71508 (A.W.) and the Gordon Family Foundation.
10. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. PMID: 10751361
11. Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95:4607–4612. PMID: 9539785
12. Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. PMID: 9893348
13. Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–165. PMID: 9916127
14. Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156:361–381. PMID: 10666364
15. Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA. 2000;97:14608–14613. PMID: 11121063
16. Baish JW, Jain RK. Fractals and cancer. Cancer Res. 2000;60:3683–3688. PMID: 10919633
17. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. PMID: 11001068
18. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. PMID: 9018236
19. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–752. PMID: 10487832
20. Zhong H, Bowen JP. Antiangiogenesis drug design: multiple pathways targeting tumor vasculature. Curr Med Chem. 2006;13:849–862. PMID: 16611071
21. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. PMID: 10647931
22. Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. PMID: 11021964
23. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. PMID: 15475116
24. Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. PMID: 15863032
26. Ray PE, Tassi E, Liu XH, Wellstein A. Role of fibroblast growth factor-binding protein in the pathogenesis of HIV-associated hemolytic uremic syndrome. Am J Physiol Regul Integr Comp Physiol. 2006;290:R105–R113. PMID: 16352855
27. Tassi E, Al-Attar A, Aigner A, Swift MR, McDonnell K, Karavanov A, et al. Enhancement of fibroblast growth factor (FGF) activity by an FGF-binding protein. J Biol Chem. 2001;276:40247–40253. PMID: 11509569
28. Mongiat M, Otto J, Oldershaw R, Ferrer F, Sato JD, Iozzo RV. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J Biol Chem. 2001;276:10263–10271. PMID: 11148217
29. Wu DQ, Kan MK, Sato GH, Okamoto T, Sato JD. Characterization and molecular cloning of a putative binding protein for heparin-binding growth factors. J Biol Chem. 1991;266:16778–16785. PMID: 1885605
30. Czubayko F, Smith RV, Chung HC, Wellstein A. Tumor growth and angiogenesis induced by a secreted binding protein for fibroblast growth factors. J Biol Chem. 1994;269:28243–28248. PMID: 7525570
31. Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med. 1997;3:1137–1140. PMID: 9334727
32. Kurtz A, Wang HL, Darwiche N, Harris V, Wellstein A. Expression of a binding protein for FGF is associated with epithelial development and skin carcinogenesis. Oncogene. 1997;14:2671–2681. PMID: 9178765
33. Xie B, Tassi E, Swift MR, McDonnell K, Bowden ET, Wang S, et al. Identification of the fibroblast growth factor (FGF)-interacting domain in a secreted FGF-binding protein by phage display. J Biol Chem. 2006;281:1137–1144. PMID: 16257968
34. McDonnell K, Bowden ET, Cabal-Manzano R, Hoxter B, Riegel AT, Wellstein A. Vascular leakage in chick embryos after expression of a secreted binding protein for fibroblast growth factors. Lab Invest. 2005;85:747–755. PMID: 15806140
35. Kagan BL, Henke RT, Cabal-Manzano R, Stoica GE, Nguyen Q, Wellstein A, et al. Complex regulation of the fibroblast growth factor-binding protein in MDA- MB-468 breast cancer cells by CCAAT/enhancer-binding protein beta. Cancer Res. 2003;63:1696–1705. PMID: 12670924
36. Ray R, Cabal-Manzano R, Moser AR, Waldman T, Zipper LM, Aigner A, et al. Up-regulation of fibroblast growth factor-binding protein, by beta-catenin during colon carcinogenesis. Cancer Res. 2003;63:8085–8089. PMID: 14678957
37. Tassi E, Henke RT, Bowden ET, Swift MR, Kodack DP, Kuo AH, et al. Expression of a fibroblast growth factor-binding protein during the development of adenocarcinoma of the pancreas and colon. Cancer Res. 2006;66:1191–1198. PMID: 16424058
38. Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA Cancer J Clin. 2001;51:15–36. PMID: 11577478
39. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. PMID: 8861899
40. Vogelstein B, Kinzler KW. The genetic basis for human cancer. 2001. 2nd edToronto: McGraw-Hill.
41. Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's 'two-hit' hypothesis. Nat Med. 1999;5:1071–1075. PMID: 10470088
42. Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54:5953–5958. PMID: 7954428
43. Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta. 1997;1332:127–147.
44. Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, et al. Association of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. PMID: 8259518
45. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734–1737. PMID: 8259519
46. Bienz M. APC: the plot thickens. Curr Opin Genet Dev. 1999;9:595–603. PMID: 10508699
47. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. PMID: 7708772
48. Gumbiner BM. Carcinogenesis: a balance between beta-catenin and APC. Curr Biol. 1997;7:R443–R446. PMID: 9210368
50. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. PMID: 9727977
51. Aoki M, Hecht A, Kruse U, Kemler R, Vogt PK. Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid-enhancing factor 1. Proc Natl Acad Sci USA. 1999;96:139–144. PMID: 9874785
52. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. PMID: 10201372
53. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. PMID: 2296722
54. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. PMID: 2453289
55. Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. PMID: 3587348
56. Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327:298–303. PMID: 2438556
57. Kressner U, Bjorheim J, Westring S, Wahlberg SS, Pahlman L, Glimelius B, et al. Ki-ras mutations and prognosis in colorectal cancer. Eur J Cancer. 1998;34:518–521. PMID: 9713302
58. Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. PMID: 2547513
59. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. PMID: 2188735
60. Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol. 2000;10:147–154. PMID: 10740269
61. Shibata D, Reale MA, Lavin P, Silverman M, Fearon ER, Steele G, et al. The DCC protein and prognosis in colorectal cancer. N Engl J Med. 1996;335:1727–1732. PMID: 8929264
62. Cho KR, Oliner JD, Simons JW, Hedrick L, Fearon ER, Preisinger AC, et al. The DCC gene: structural analysis and mutations in colorectal carcinomas. Genomics. 1994;19:525–531. PMID: 8188295
63. Chen YQ, Hsieh JT, Yao F, Fang B, Pong RC, Cipriano SC, et al. Induction of apoptosis and G2/M cell cycle arrest by DCC. Oncogene. 1999;18:2747–2754. PMID: 10348349
64. Tarafa G, Villanueva A, Farre L, Rodriguez J, Musulen E, Reyes G, et al. DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene. 2000;19:546–555. PMID: 10698524
65. Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098–3103. PMID: 10340381
66. Koyama M, Ito M, Nagai H, Emi M, Moriyama Y. Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: identification of seven novel somatic mutations in tumors from Japanese patients. Mutat Res. 1999;406:71–77. PMID: 10479724
67. Salovaara R, Roth S, Loukola A, Launonen V, Sistonen P, Avizienyte E, et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002;51:56–59. PMID: 12077092
68. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectaltumor development. N Engl J Med. 1988;319:525–532. PMID: 2841597
69. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. PMID: 2649981
70. Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. PMID: 10430607
71. Zeng ZS, Sarkis AS, Zhang ZF, Klimstra DS, Charytonowicz E, Guillem JG, et al. p53 nuclear overexpression: an independent predictor of survival in lymph node--positive colorectal cancer patients. J Clin Oncol. 1994;12:2043–2050. PMID: 7931472
72. Leahy DT, Salman R, Mulcahy H, Sheahan K, O'Donoghue DP, Parfrey NA. Prognostic significance of p53 abnormalities in colorectal carcinoma detected by PCR-SSCP and immunohistochemical analysis. J Pathol. 1996;180:364–370. PMID: 9014855
73. Allegra CJ, Paik S, Colangelo LH, Parr AL, Kirsch I, Kim G, et al. Prognostic value of thymidylate synthase, Ki-67, and p53 in patients with Dukes' B and C colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project collaborative study. J Clin Oncol. 2003;21:241–250. PMID: 12525515
74. Garrity MM, Burgart LJ, Mahoney MR, Windschitl HE, Salim M, Wiesenfeld M, et al. Prognostic value of proliferation, apoptosis, defective DNA mismatch repair, and p53 overexpression in patients with resected Dukes' B2 or C colon cancer: a North Central Cancer Treatment Group Study. J Clin Oncol. 2004;22:1572–1582. PMID: 15117979
75. Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. PMID: 15661684
76. Bodner WR, Hilaris BS, Mastoras DA. Radiation therapy in pancreatic cancer: current practice and future trends. J Clin Gastroenterol. 2000;30:230–233. PMID: 10777178
77. Berlin JD, Rothenberg M. Chemotherapy for resectable and advanced pancreatic cancer. Oncology. 2001;15:1241–1249. PMID: 11702956
78. Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. PMID: 11342768
79. Sessa F, Solcia E, Capella C, Bonato M, Scarpa A, Zamboni G, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch. 1994;425:357–367. PMID: 7820300
80. Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. PMID: 10854204
81. Hruban RH, Offerhaus GJ, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-suppressor genes in pancreatic cancer. J Hepatobiliary Pancreat Surg. 1998;5:383–391. PMID: 9931387
82. Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8PMID: 12556241
83. Siddiqi I, Funatomi H, Kobrin MS, Friess H, Buchler MW, Korc M. Increased expression of keratinocyte growth factor in human pancreatic cancer. Biochem Biophys Res Commun. 1995;215:309–315. PMID: 7575607
84. Kuwahara K, Sasaki T, Kuwada Y, Murakami M, Yamasaki S, Chayama K. Expressions of angiogenic factors in pancreatic ductal carcinoma: a correlative study with clinicopathologic parameters and patient survival. Pancreas. 2003;26:344–349. PMID: 12717266
85. Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, et al. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res. 1993;53:5289–5296. PMID: 7693336
86. Kornmann M, Ishiwata T, Beger HG, Korc M. Fibroblast growth factor-5 stimulates mitogenic signaling and is overexpressed in human pancreatic cancer: evidence for autocrine and paracrine actions. Oncogene. 1997;15:1417–1424. PMID: 9333017
87. Kleeff J, Kothari NH, Friess H, Fan H, Korc M. Adenovirus-mediated transfer of a truncated fibroblast growth factor (FGF) type I receptor blocks FGF-2 signaling in multiple pancreatic cancer cell lines. Pancreas. 2004;28:25–30. PMID: 14707726
88. Kornmann M, Ishiwata T, Matsuda K, Lopez ME, Fukahi K, Asano G, et al. IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology. 2002;123:301–313. PMID: 12105858
89. Kornmann M, Beger HG, Korc M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas. 1998;17:169–175. PMID: 9700949
90. Wagner M, Lopez ME, Cahn M, Korc M. Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 1998;114:798–807. PMID: 9516401
91. Wray CJ, Rilo HL, Ahmad SA. Colon cancer angiogenesis and antiangiogenic therapy. Expert Opin Investig Drugs. 2004;13:631–641.
92. Mancuso A, Sternberg CN. Colorectal cancer and antiangiogenic therapy: what can be expected in clinical practice? Crit Rev Oncol Hematol. 2005;55:67–81. PMID: 15890525
93. Dirix LY, Vermeulen PB, Hubens G, Benoy I, Martin M, De Pooter C, et al. Serum basic fibroblast growth factor and vascular endothelial growth factor and tumour growth kinetics in advanced colorectal cancer. Ann Oncol. 1996;7:843–848. PMID: 8922199
94. Landriscina M, Cassano A, Ratto C, Longo R, Ippoliti M, Palazzotti B, et al. Quantitative analysis of basic fibroblast growth factor and vascular endothelial growth factor in human colorectal cancer. Br J Cancer. 1998;78:765–770. PMID: 9743297
95. Galzie Z, Fernig DG, Smith JA, Poston GJ, Kinsella AR. Invasion of human colorectal carcinoma cells is promoted by endogenous basic fibroblast growth factor. Int J Cancer. 1997;71:390–395. PMID: 9139874
96. Stoeltzing O, Liu W, Reinmuth N, Parikh A, Ahmad SA, Jung YD, et al. Angiogenesis and antiangiogenic therapy of colon cancer liver metastasis. Ann Surg Oncol. 2003;10:722–733. PMID: 12900362
97. Tahara E. In : Meyers RA, editor. Growth factors and oncogenes in gastrointestinal cancers. Encyclopedia of molecular cell biology and molecular medicine. 2005. Weinheim: Wiley-VCH Verlag GmbH & Co KGaA; p. 1–31.
98. Akbulut H, Altuntas F, Akbulut KG, Ozturk G, Cindoruk M, Unal E, et al. Prognostic role of serum vascular endothelial growth factor, basic fibroblast growth factor and nitric oxide in patients with colorectal carcinoma. Cytokine. 2002;20:184–190. PMID: 12543084
99. Herfarth H, Brand K, Rath HC, Rogler G, Scholmerich J, Falk W. Nuclear factor-kappa B activity and intestinal inflammation in dextran sulphate sodium (DSS)-induced colitis in mice is suppressed by gliotoxin. Clin Exp Immunol. 2000;120:59–65. PMID: 10759764
100. Giavazzi R, Albini A, Bussolino F, DeBraud F, Presta M, Ziche M, et al. The biological basis for antiangiogenic therapy. Eur J Cancer. 2000;36:1913–1918. PMID: 11000570
101. Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21:505–515. PMID: 10688871
102. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. PMID: 15637262
103. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. PMID: 15175435
104. Kabbinavar FF, Schulz J, McCleod M, Patel T, Hamm JT, Hecht JR, et al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol. 2005;23:3697–3705. PMID: 15738537
105. de Castro Junior G, Puglisi F, de Azambuja E, El Saghir NS, Awada A. Angiogenesis and cancer: a cross-talk between basic science and clinical trials (the "do ut des" paradigm). Crit Rev Oncol Hematol. 2006;59:40–50. PMID: 16600618
106. Cao Y. Tumor angiogenesis and therapy. Biomed Pharmacother. 2005;59(Suppl 2):S340–S343. PMID: 16507405
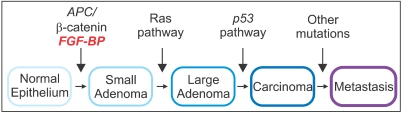
Fig. 1
Genetic alterations during the development of colorectal cancer. Significant genetic alterations at different junctures during the transformation of colon epithelia to invasive adenocarcinoma are depicted. Induction of FGF-BP expression is an early event that is driven by mutations and activation of the WNT/beta-catenin pathway (Adapted and modified from Tassi E. and Wellstein A., Sem. Onc., in press).
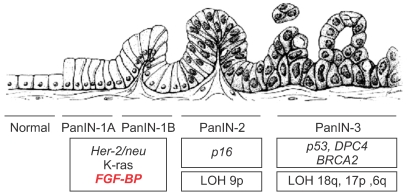
Fig. 2
Genetic alterations during malignant transformation of pancreas epithelia. The progression from normal duct epithelium to low-grade and high-grade PanIN (Pancreatic Intraepithelial Neoplasia) and the associated accumulation of genetic alterations are shown (Adapted and modified from Tassi E. and Wellstein A., Sem. Onc., in press).
Fig. 3
FGF-BP expression in human and mouse dysplastic lesions. (A) Frequency of FGF-BP protein expression, analyzed by immunohistochemistry (IHC) with a polyclonal anti-FGF-BP antibody in 161 human specimens ***P<0.0001. (B) Frequency of FGF-BP mRNA expression in normal and dysplastic intestinal tissues obtained from B6 APCMin/+ mice. FGF-BP expression coincides with the activation of the beta-catenin pathway in this model **P<0.001 (36) (Adapted and modified from Tassi E. and Wellstein A., Sem. Onc., in press).
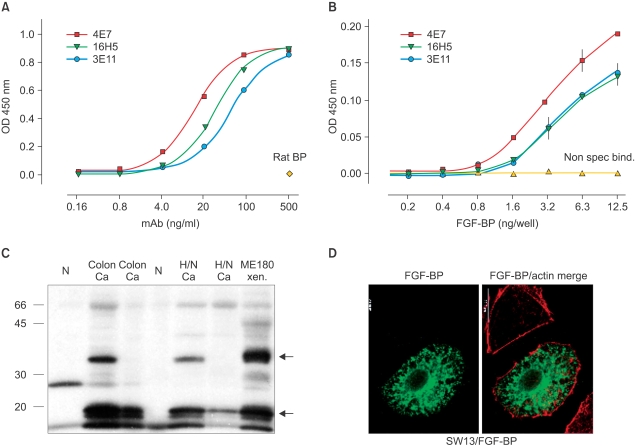
Fig. 4
Characterization of FGF-BP monoclonal antibodies. (A) and (B) ELISA assay with mouse monoclonal antibodies to human FGF-BP. (A) specificity of the mAbs. Recombinant FGF-BP (50 ng/well) was immobilized in a microtiter plate and detected with different concentrations of mAbs indicated. (B) sensitivity of the mAb. Different concentrations of recombinant FGF-BP were immobilized and incubated with 0.2µg/ml of the mAbs indicated. (C) Western blot analysis of FGF-BP from frozen surgical samples of colon and head and neck (H/N) cancer tissues and ME180 xenografts. N=normal tissue. (D) Immunofluorescence of FGF-BP transiently transfected SW13 cells. Only some of SW13 transfected cells express detectable FGF-BP protein (green) and the F-actin stain highlights all cells (red) (Adapted and modified from (37)).
Fig. 5
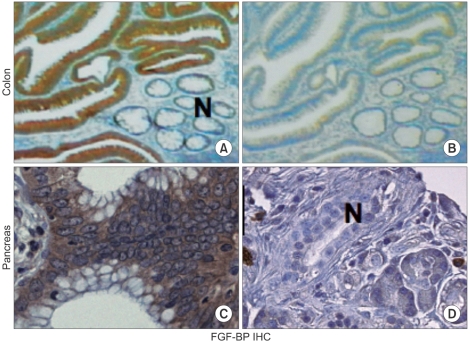
FGF-BP staining in premalignant human colon and pancreas lesions. (A) Histochemistry with a monoclonal antibody against FGF-BP shows expression (dark brown staining) in dysplastic lesions of the colon next to normal crypt epithelia (N). (B) A serial section from panel A stained with the secondary antibody only. (C) FGF-BP staining in a PanIN lesion. (D) A normal pancreatic duct shows no staining for FGF-BP. Further details in Ref. (37) (Adapted and modified from Tassi E. and Wellstein A., Sem. Onc., in press).
Fig. 6
Growth inhibition by a monoclonal Ab to FGF-BP. Soft agar colony formation of SW13/FGF-BP or mock transfected SW13 cells treated with the 4E7 mAb (40µg/ml). The number of colonies is given in the abscence and presence of FGF-2±the mAb (Adapted and modified from (37)).