Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the digestive tract. Most GISTs harbor activating mutations of the genes encoding KIT or platelet-derived growth factor receptor alpha (PDGFRA), resulting in the constitutive activation of protein tyrosine kinase signaling [1]. KIT mutations occur in 70%-85% of GISTs and are most common in exon 11, followed by exon 9, whereas exon 13 and 17 mutations are rare. PDGFRA mutations are detected in 5%-15% of GISTs and are mutually exclusive to KIT mutations [2-4].
Imatinib (Glivec/Gleevec, Novartis Oncology, East Hanover, NJ) has shown remarkably enhanced efficacy over conventional chemotherapy and has become the standard first-line treatment for patients with unresectable or metastatic GISTs [5-8]. Extensive correlative analyses have identified the types of KIT mutations associated with the efficacy of imatinib in GISTs [9-11]: with imatinib treatment patients with KIT exon 9 mutations show poorer outcomes than those with KIT exon 11 mutations.
The association between imatinib sensitivity and PDGFRA mutations was investigated in a subgroup of patients included in prospective clinical trials, and in vitro and in vivo studies reported association of the PDGFRA exon 18 D842V substitution with resistance to imatinib [9,10,12]. However, because of the low incidence of PDGFRA mutations in advanced GISTs, data on the activity of imatinib against PDGFRA-mutant GISTs were mostly obtained in a small number of patients.
A recent retrospective study conducted in Europe analyzed 55 patients with PDGFRA-mutant GISTs [13]. Although this study is the largest of its kind to date, additional data are still needed in order to improve our understanding of these types of mutations, particularly considering the small sample size of previous studies. Therefore, we retrospectively analyzed the efficacy of first-line imatinib in patients with advanced GISTs harboring PDGFRA mutations.
Materials and Methods
Patients with PDGFRA-mutant GISTs who received imatinib as primary therapy for advanced disease between January 2000 and June 2012 were identified from the GIST registry of Asan Medical Center, Seoul, Korea. Data were collected from both the registry and retrospectively by review of medical records. During imatinib treatment, all patients were monitored regularly for evaluation of response by computed tomography scan every 3 months. Magnetic resonance imaging or positron emission tomography was performed when necessary. Response was assessed according to the Response Evaluation Criteria in Solid Tumors ver. 1.0 with some modifications [14]. Lesions showing necrosis, despite their increased size, and new cystic lesions in the liver were not considered disease progression [15]. This study was approved by the Institutional Review Board of Asan Medical Center.
For mutational analyses of the KIT and PDGFRA genes, 10 μm tumor sections were cut from formalin-fixed paraffinembedded tissue samples. Genomic DNA was extracted from three sections using a DEXPAT KIT (TaKaRa, Kyoto, Japan). Exons 9, 11, 13, and 17 of KIT, and exons 12 and 18 of PDGFRA were amplified by polymerase chain reaction (PCR). The PCR products were sub-cloned using a TOPO TA cloning KIT (K4500-01, Invitrogen, Carlsbad, CA) and sequenced twice using an ABI 3700 DNA analyzer (Applied Biosystems, Foster City, CA).
The chi-square test or Fisher exact test was used to assess the association between categorical variables, as appropriate. The probability of survival was estimated using the Kaplan-Meier method and compared using the log-rank test. Progression-free survival (PFS) was defined as the time from the first dose of imatinib to disease progression or death from any cause. Overall survival (OS) was estimated as the time from the first dose of imatinib until death due to any cause, and censored at the last follow-up visit for patients who were still alive. Statistical analyses were performed using the SPSS ver. 18.0 (SPSS Inc., Chicago, IL). All tests were two-sided, with 5% serving as the level of significance.
Results
1. Patient characteristics
A total of 823 patients were genotyped for KIT and PDGFRA. PDGFRA mutations were detected in 35 patients (4%), and 19 patients (2%) received imatinib as first-line therapy for advanced disease. After exclusion of one patient who had received adjuvant imatinib prior to the use of imatinib for recurrent disease, 18 patients were analyzed to estimate the sensitivity of PDGFRA-mutant GISTs to imatinib. The patients’ baseline characteristics are shown in Table 1. The patients’ median age at the start of imatinib therapy was 58 years (range, 31 to 74 years) and 11 patients (61%) were male. Most patients in our series (n=17, 94%) were KIT expression-positive by immunohistochemistry; however, weak and focal expression was noted in three cases (17%) and one case (6%), respectively. Nine patients (50%) had exon 18 D842V substitution and four patients (22%) had non-D842V exon 18 mutations. Five patients (28%) had exon 12 mutations. Detailed clinical information of all patients is summarized in Table 2.
2. Efficacy of imatinib as first-line therapy
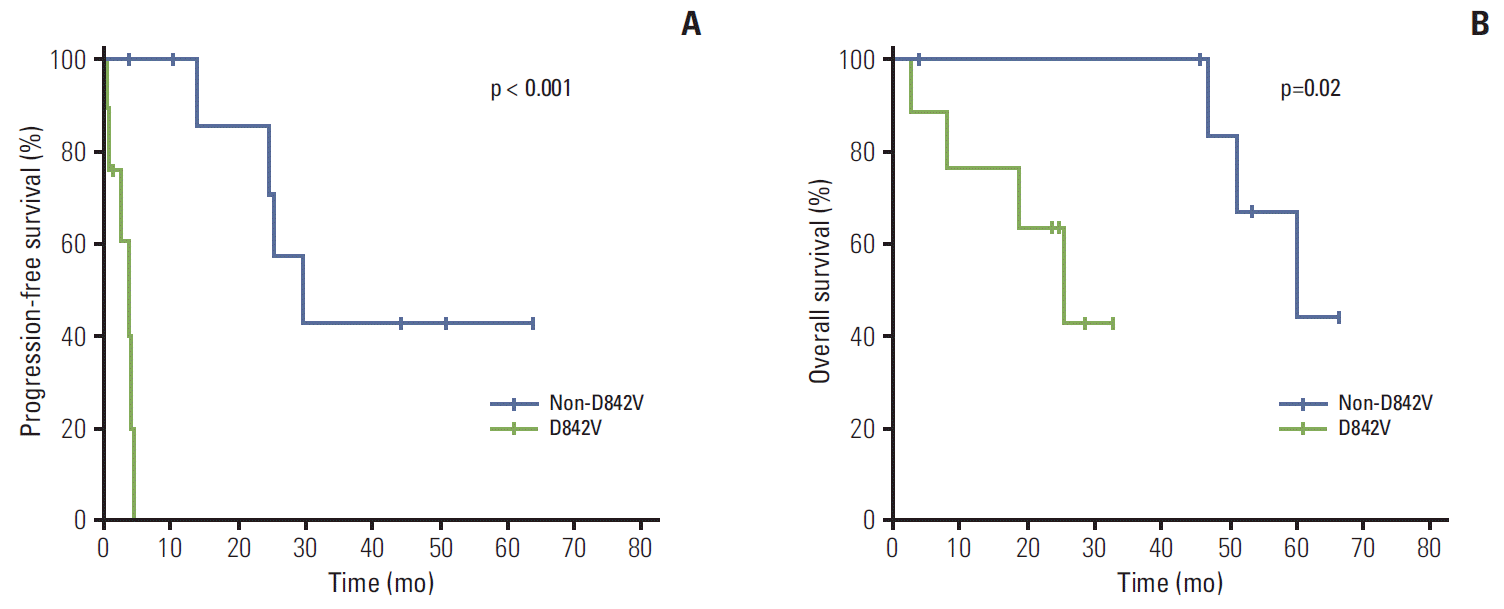
The responses to imatinib in patients with PDGFRA-mutant GISTs are summarized in Table 3. Twelve patients (67%) were available for response assessment; four had no measurable lesion after surgical resection and two were lost to follow-up before the first response evaluation. None of the patients showed a complete response; five patients (42%) showed a partial response and three patients (25%) showed stable disease. None of the five patients with the D842V mutation achieved an objective response, whereas five of seven patients (71%) with non-D842V PDGFRA mutations showed a partial response (p=0.03). With a median follow-up period of 32.5 months (range, 2.8 to 65.6 months) in surviving patients, the median PFS and OS was 24.8 months (95% confidence interval [CI], 0.0 to 57.2) and 51.2 months (95% CI, 37.1 to 65.3), respectively. Patients with D842V-mutant GISTs had significantly shorter median PFS than those with non-D842V PDGFRA-mutant GISTs: 3.8 months (95% CI, 1.4 to 6.3) vs. 29.5 months (95% CI, 18.3 to 40.7) (p < 0.001) (Fig. 1). In addition, patients with D842V-mutant GISTs showed inferior OS compared to those with non-D842V PDGFRA-mutant GISTs (median, 25.2 months; 95% CI, 12.7 to 37.8 vs. median, 59.8 months; 95% CI, 43.0 to 76.5; p=0.02).
3. Outcomes with second-line therapy
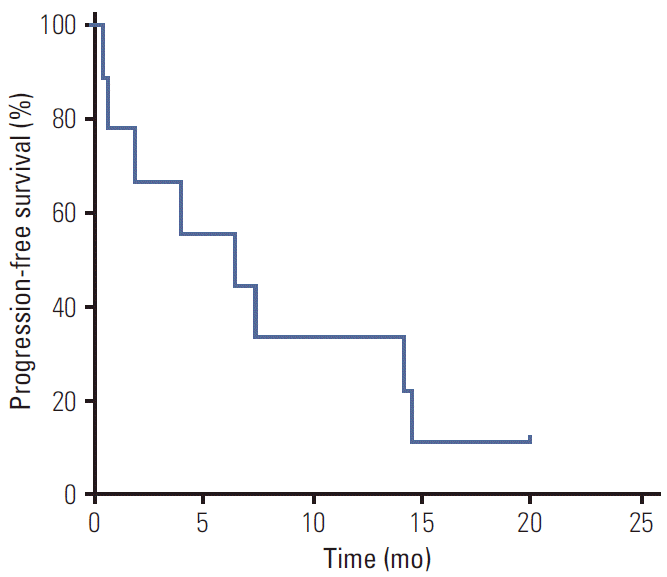
Among ten patients with documented tumor progression on a standard dose of imatinib, five (three with non-D842V mutations and two with D842V substitution) received an increased dose of imatinib (600 mg/day or 800 mg/day). None of the patients showed an objective response and the median PFS was 1.5 months (95% CI, 0.5 to 2.5). Nine patients were treated with sunitinib after failure of imatinib (three had non-D842V mutations and six had D842V substitution). The responses to second-line sunitinib treatment in the overall population and according to mutation type are summarized in Table 4. None of the patients achieved an objective response. The median PFS with second-line therapy was 6.4 months (95% CI, 0.0 to 13.5) (Fig. 2). Patients with the D842V substitution tended to show poorer PFS than those with non-D842V PDGFRAmutations (median, 1.9 months; 95% CI, 0.0 to 5.9 vs. median, 7.3 months; 95% CI, 5.9 to 8.8; p=0.26), although the difference did not reach statistical significance.
Discussion
In management of unresectable or metastatic advanced GISTs, tumor genotype, particularly KIT exon mutations, has been regarded as an important predictive factor for the efficacy of imatinib. However, because PDGFRA-mutant GISTs are generally indolent and localized with a low-risk of recurrence [1-4], data on the response to imatinib in patients with advanced disease harboring these specific mutations have been mainly based on a few cases. A relatively large European retrospective study recently reported that imatinib has little efficacy in the subgroup of patients with PDGFRA exon 18 D842V substitution, while other types of PDGFRA mutations appear to be sensitive to imatinib; however, additional data were still needed in order to improve our understanding of imatinib sensitivity in this group of patients [13]. The results of the current study are in agreement with those of previous studies, and could add to the evidence in regard to sensitivity to imatinib in PDGFRA-mutant GISTs.
In our series, PDGFRA mutations were detected in 4% of patients with GISTs genotyped for KIT and PDGFRA. These results are similar to those of two Western phase III trials [9,10] and a Korean retrospective study [16] of first-line imatinib treatment, which reported frequency of 2%-5%. However, our results differ significantly from those of previous population-based studies reporting 14%-16% as the frequency of PDGFRA mutations [2-4]. The large discrepancies in the results between population-based studies and clinical trials of imatinib could be associated with the proportion of patients with localized disease among the whole study population, considering that PDGFRA-mutant GISTs are more frequent in localized disease than in metastatic disease [2-4]. Most patients in our registry were referred from other hospitals following diagnosis. Therefore, compared with population-based studies, it is more likely that our registry included patients with high-risk or advanced disease. This might explain why the frequency of PDGFRA mutations in our study was similar to that reported in clinical trials for advanced GISTs rather than population-based studies. Most PDGFRA mutations occurred in gastric GISTs (88%), and exon 18 D842V substitution (50%) was the most common type of PDGFRA mutation, as previously reported [1]. PDGFRA mutations were more common in KIT-negative GISTs [17]. However, most of our patients (17 of 18, 94%) had KIT-positive GISTs by immunohistochemistry, although 23% had weak or focal expression. Our results are consistent with those of the European study, which included a large patient cohort, and reported that 85% of patients were KIT-positive [15]. These findings suggest that KIT is overexpressed in most PDGFRA-mutant GISTs [15] and, although mutations of KIT and PDGFRA are mutually exclusive in GISTs, expression of those proteins is not [18].
Consistent with previous preclinical and clinical studies [9-13], our results showed that GISTs with the D842V mutation are primarily resistant to imatinib. Patients with D842V mutant GISTs showed no objective response to first-line imatinib and most of them progressed within 6 months of treatment. The median PFS of this group was 3.8 months, consistent with the results of a previous retrospective study (2.8 months) [13]. This inherent resistance of D842V-mutated GISTs to imatinib can be explained as this substitution results in a change in the kinase activation loop that induces the protein conformation to favor the active conformation, considering that imatinib can only bind to the inactive conformation of PDGFRA [19]. Sustained tumor control with imatinib was previously reported in some patients [9,13]; however, there is no biomarker to differentiate patients who might potentially benefit from imatinib treatment from those who would not. Because of the lack of efficacy of imatinib in patients with this mutation, mutational analysis should be performed to exclude GIST patients with the PDGFRA D842V substitution before administration of adjuvant imatinib after curative surgical resection or neoadjuvant imatinib for organ preservation or marginally resectable disease [20].
For patients with metastatic or unresectable GISTs with D842V mutation, a phase 2 study of crenolanib, a novel platelet-derived growth factor receptor inhibitor, is ongoing for this subgroup of patients based on the promising preclinical activity (NCT01243346) [21]; however, there is no currently approved drug with activity against the D842V mutation [12,22]. Therefore, these patients should be encouraged to participate in clinical trials for investigational drugs with potential activity against this mutation. If such drugs are not an option, imatinib therapy should be tried for the potential chance of achieving disease control.
In addition to the D842V substitution, in-frame deletion, deletion/insertion and substitutions in PDGFRA exon 18, and deletions, substitutions and duplications in exon 12 have been previously identified [1]. Although rare, substitutions in exon 4 and 14 have also been reported [1,2,13]. In the current study, patients with non-D842V mutant GISTs had various mutations in exons 18 and 12. In line with previous in vitro studies investigating the sensitivity of imatinib against non-D842V PDGFRA mutations [1,12], first-line imatinib was effective in our GIST patients with non-D842V PDGFRA mutations, who showed a median PFS of 29.5 months. This finding is consistent with the results of a previous analysis of non-D842V PDGFRA-mutant GISTs, which showed association with superior PFS and OS compared to D842V mutant GISTs [13]. Compared with the outcomes of patients with different genotypes in previous studies, PFS of our patients with non-D842V PDGFRA mutations was similar to that previously reported for patients with KIT exon 11 mutations in Western trials [9-11], and those with KIT exon 9 mutations in a Korean multicenter retrospective analysis [16], suggesting that sensitivity to imatinib in this patient subgroup may be similar to that of patients with KITmutations. In the current study, one patient with Del D842 showed long-term disease control with partial response, and progressed after approximately 2 years of imatinib therapy. A previous study that included two patients with Del D842 reported that one patient showed a long-term response [13]. Taken together with the in vitro activity of imatinib in GISTs with D842-involving deletions [12,23], these findings suggest that the nature of the mutation, in addition to its location, plays an important role in the sensitivity to imatinib, in addition to its location [13].
The current study had several limitations associated with its retrospective nature and the small sample size. However, PDGFRAmutations are infrequent, particularly in advanced disease, and few studies have investigated the therapeutic outcomes of this subgroup. Therefore, our results may provide guidance in the treatment of patients with GISTs harboring PDGFRA mutations.
Conclusion
We showed that imatinib is active against non-D842V PDGFRA-mutant GISTs, whereas GISTs harboring the D842V mutation are primarily resistant to imatinib. Considering that no approved drug has shown efficacy against D842V-mutant GISTs, enrollment in clinical trials should be encouraged in this group of patients, especially if investigational drugs with in vitro activity are identified.